
REGULATION OF ARTERIAL STIFFNESS: CELLULAR, MOLECULAR AND NEUROGENIC MECHANISMS☆
Presented as the invited McDonald Lecture by Alberto Avolio at the Artery11 Meeting of the Association for Research into Arterial Structure and Physiology, Maison Internationale, Cité Universitaire, Paris, France, 13–15 October, 2011.
- DOI
- 10.1016/j.artres.2011.10.002How to use a DOI?
- Keywords
- Pulse wave velocity; Extracellular matrix; Vascular smooth muscle; Calcification; Post-translational modification; S-nitrosylation; Tissue transglutaminase
- Abstract
The lasting legacy of Donald McDonald has been in the establishment of physiological and biophysical principles of the relation of blood pressure and flow in arteries. This relation is determined by physical properties of arteries, among which wall stiffness is a dominant parameter. Increased arterial stiffness leads to an increase in pulse pressure due to alterations in the capacitive properties of large arteries and the increase in pulse wave velocity, which leads to early return of reflected waves. While the haemodynamic and biophysical effects of arterial stiffness have been studied extensively and are well established, the underlying mechanisms responsible for the alteration of the structural properties of the arterial wall are not as well understood. Some potential mechanisms will be addressed in relation to the interaction of the cellular and acellular components and their effect on the structural integrity of the arterial wall. The modification of the smooth muscle cell to influence medial calcification and the endothelium-dependent nitric oxide pathways affecting the extracellular matrix through post-translational modification of proteins form part of positive feedback mechanisms in the regulation of arterial stiffness through cellular and molecular processes. This is further modulated by neurogenic effects on smooth muscle contractility affecting wall stiffness. While the passive effects on the arterial wall due to blood pressure and heart rate cannot be readily modified, uncovering cellular, molecular and neurogenic mechanisms regulating arterial stiffness can offer novel means to interrogate pathways leading to the detrimental effects of degeneration of arterial function and altered relation of pressure and flow.
- Copyright
- © 2011 Association for Research into Arterial Structure and Physiology. Physiology. Published by Elsevier B.V. All rights reserved.
- Open Access
- This is an open access article distributed under the CC BY-NC license.
Introduction
The elasticity of arteries is a fundamental determinant of the relationship between pulsatile pressure and flow.1 The characterisation of pressure–flow relationships either in the time or frequency domain enables the quantification of the hydraulic load such that it can describe the impedance to flow due to the physical properties of arteries. The loss of elasticity of the artery wall leads to stiffening of the conduit vessels, reducing their buffering capacity as well as increasing the speed of the propagating pulse along the vessel wall. Hence, for a given ventricular stroke volume, arterial stiffness is a major determinant of pulse pressure due to the combined influence on the capacitive effects of the artery wall to absorb the pulsatile energy and the wave propagation effects that influence peripheral wave reflection. It is these factors that are integral to the underlying mechanisms responsible for the gradual increase in systolic pressure with age leading to development of isolated systolic hypertension in the elderly and increased cardiovascular risk.2–4 It is also these mechanisms that have a dominant role in the significance of pulse pressure5 and the emergence of arterial pulse wave velocity as an increasingly powerful independent predictor of cardiovascular morbidity and mortality6–8 and significant reclassifier of cardiovascular risk.9
Donald McDonald, during his professional life as an investigative physiologist and with the early collaborations with John Womersley and Michael Taylor, played a highly decisive role in the establishment of the field of haemodynamics on which the above mechanistic explanations are based.10–12 His work, and that of many others that aimed to describe the pressure/flow relationships in a closed form such that fundamental characteristics of experimental measurements could be elucidated by empirical models, laid the foundations for the now largely accepted understanding of the effects of arterial structure and function on the haemodynamic determinants of pulsatile blood flow in arteries.1 The field has now matured and evolved into investigations of flow-dependent effects on the arterial properties themselves.13,14 Hence, while biophysical foundations of the functional haemodynamic elements are well established, the mechanisms of what alter physical properties such as arterial stiffness are not as well understood.
The combination of the biophysical principles in arterial haemodynamics with the complex but powerful emerging field of molecular biology in the recent two decades is enabling investigations into the underlying factors that translate structural changes and modifications of artery wall constituents to functional correlates. These seen as increased pulse wave velocity and arterial pulse pressure, both highly significant factors of cardiovascular risk and end organ damage. The artery wall constituents can be altered by passive stimuli, such as increased mechanical stress due to distending pressure. These lead to structural disorganization, fatiguing effects and fragmentation of elastic fibres. Alterations can also result from active changes mediated through a cascade of biochemical cellular signaling processes affecting the integrity of the extracellular matrix, translating to altered arterial functional properties. Molecular probes are making it possible to uncover pathways involved in the interaction between cellular processes and extracellular matrix in the artery wall15 through biochemical and mechanotransduction signaling,16,17 thus opening up avenues for active interrogation of these pathways for direct regulation of arterial stiffness.
Cellular and molecular mechanisms
The cellular components of the artery wall are now accepted as providing the active machinery for maintaining homeostatic balance. Thus, dysregulation of cellular processes will inevitably affect the local milieu and the ensuing mechanical properties. Vascular smooth muscle (VSM) cells can alter their phenotype from performing principally a contractile function to a synthetic function. They can also undergo transdifferentiation into osteoblasts leading to mineralization and calcium deposition in the arterial media,18 with direct consequences on arterial stiffness.19,20 Endothelial cells, while once thought to constitute an inactive lining of the vessel lumen, are now recognized as being an integral part of the regulatory mechanisms for flow-dependent properties of vascular function through the nitric oxide pathways.21
Post-translational modification of proteins; S-nitrosylation, tissue tranglutaminase and arterial stiffness
The effect of cellular biochemistry on the arterial wall is through the cascade of events leading to protein modification. Driven by rapid advances in proteomic analysis,22 there has been recent interest in the description of protein modifications underlying cardiovascular biology. Indeed, with increasing proteomic data, there is increasing evidence that post-translation modification of proteins is a common occurrence. For example, more that 60% of the known cardiac proteins undergo some form of post-translational modification, with the most common being phosphorylation.22
Nitric oxide (NO) has been classified as the “third respiratory gas”,23 and since its discovery it is being increasingly recognized as having a ubiquitous effect on cellular and acellular processes. One of the first receptors to be identified for NO was soluble guanylyl cyclase, producing cyclic guanosine monophosphate, (cGMP), which potentiates VSM relaxation.24,25 However, it is now becoming apparent that a large part of the NO influence may be through cGMP independent pathways, such as the post-translational modification process of S-nitrosylation. In S-nitrosylation, a protein cysteine thiol undergoes covalent modification by an NO group and generates an S-nitrosothiol (SNO).23
The S-nitrosylation process of the tissue transglutminase protein type 2 (TG2) has been shown to be involved in the calcium-dependent TG2 mediated modification of the vascular extracellular matrix through formation of collagen cross-linking, affecting wall stiffness.26 TG2, a form of ‘biological glue’27 is one of a family of transglutmanises, enzymes that form stable structures.28,29 The endothelial production of NO produces a cyclic redox-dependent S-nitrosylation and denitrosylation of TG2.23 The reduced S-nitrosylation (and therefore increased denitrosylation) of TG2 that takes place with reduced production or bioavailability of NO (eg due to endothelial dysfunction) causes exteriorization of the protein to the extracellular space. Increased activity of matrix TG2 has been shown to be associated with increased aortic pulse wave velocity in TG2 knockout mouse models.30
Recent studies in ageing rats and TG2 and eNOS knockout mice models have shown that reduction of bioavailability of NO as occurs with ageing, inflammation and endothelial dysfunction in general is associated with cellular mechanisms contributing to arterial stiffness.31 Many other studies have also shown that mechanical shear forces on the endothelial cells are critical in modulating the expression of endothelial NO synthase (eNOS) and gene expression.13,14,32 In vitro perfusion studies of endothelial cells cultured in compliant tubes indicate that the stiffness of the tube material can cause changes in eNOS production.32 If this mechanism can be shown to be active over the range of stiffness changes found in vivo, it suggests a potentially powerful positive feedback system, where stimuli that lead to a reduction in NO production resulting in increased wall stiffness form part of a cascade where increased wall stiffness then leads to further reduction of NO (Fig. 1).
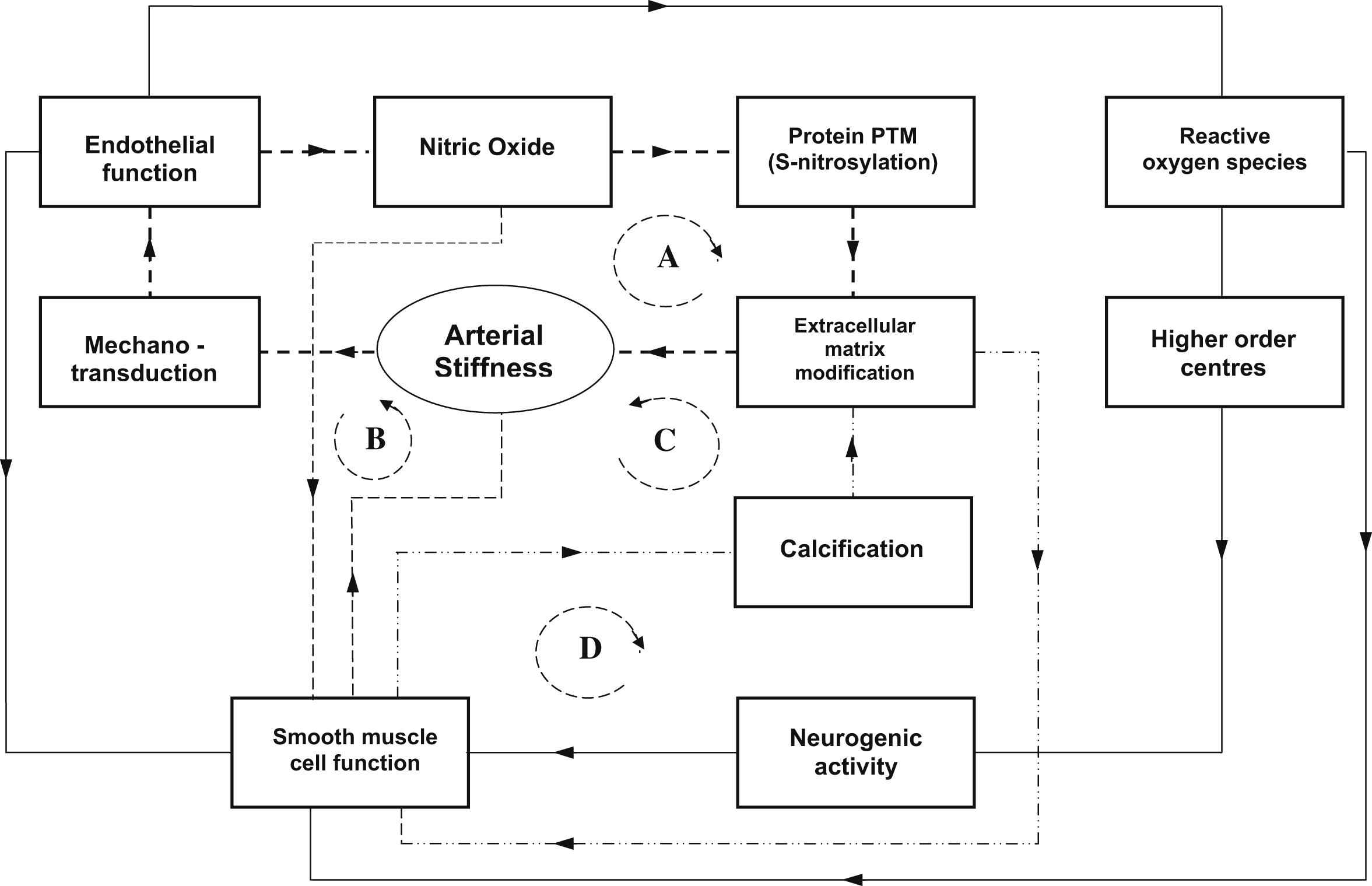
Schematic diagram illustrating the possible interaction between the cellular, molecular and neurogenic influences on arterial stiffness. The paths indicated by dotted lines indicate effective positive feedback loops (A,B,C,D) where the effect on arterial stiffness is amplified. A. Loss of endothelial function reduces nitric oxide (NO) bioavailability causing changes in protein post-translational modification (eg reduced S-nitrosylation of TG2) and increased cross-linking in extracellular matrix resulting in increased arterial stiffness.15 Increased arterial stiffness further reduces endothelial function via mechanotransduction effects.32 B. The direct effects of vascular smooth muscle (VSM) on arterial stiffness due to external signals (eg neurogenic interaction with parts of path A). Reduced NO has the effect of increasing VSM tone. In conditions where increased VSM tone increases wall stiffness, the effect is amplified via path A mechanisms. In conditions where increased VSM tone leads to reduced stiffness, this becomes a negative feedback loop that tends to stabilize the system. C. VSM cell transdifferention leads to calcification and increasing arterial stiffness through extracellular matrix modification which alters extracellular matrix due to calcification. Positive feedback occurs through paths B and C. D. Changes in millieu of extracellular matrix (eg elastin frangmentation, TG2) potentiates VSM changes of phenotype providing positive feedback also for path C. Function of endothelial cells is also a determinant of VSM cell function,61 which is modulated by neurogenic inputs. Recent studies suggest a potential connection between endothelial function and downstream neurogenic activity via reactive oxygen species (ROS) in the brain affecting sympathetic activity62 which maintains VSM enervation.
Vascular smooth muscle cells
The effects of smooth muscle cells on the stiffness of the artery wall depend on the location in the arterial tree and the relative amount of VSM content. The effect is mediated through the contractile state of the VSM cell. Experiments have quantified the intrinsic effect of VSM on the elastic modulus of the arterial wall.1 Activation of VSM affects the viscoelastic properties of the arterial wall and so the overall frequency dependency of the elastic modulus.33,34 Tensile testing studies of activated and deactivated VSM suggest that at a given level of resting strain, contraction of VSM is associated with a lower elastic modulus. Conversely a relaxed artery becomes stiffer. The explanation for these seemingly paradoxical observations is that with VSM contraction, more of the load is borne by the elastin fibres and less by the collagen fibres. The converse is true with VSM relaxation.35,36
VSM and arterial calcification
Calcification of the media of the artery wall is associated with increased arterial stiffness.19,20,37 The principle stimulus for arterial calcification is now accepted as being the change of phenotype of the VSM cell from one which responds to activation to one that mediates a type of bone formation.38 Early interpretation of ectopic mineralization occurring in blood vessels was assumed to be related to the passive precipitation of calcium and phosphate. However, it is now accepted as being a highly regulated active process where calcium deposition occurs via osteoblast-like cells which are principally derived from existing VSM cells, as well as from stem cells present in the circulation or in the wall of the vessel.18,38 The process of cellular transdifferentiation is induced by a number of factors associated with degenerative changes to the extracellular matrix such as degradation of elastin fibres, fibrosis, vitamin D and oxidative stress.18,38 Since elastin degradation is associated with arterial stiffness, this is a positive feedback path where wall stiffness is further increased via wall calcification through VSM differentiation (Fig. 1). This disturbs the normal balance that exists between promotion and inhibition of calcification, with the disturbance being increased with age and chronic conditions such as kidney disease and diabetes,38 conditions that are also associated with increased arterial stiffness.39
Arterial calcification and TG2
In addition to the effect on extracellular matrix, TG2 has been shown to affect the transdifferentiation of VSM cells that leads to arterial calcification.40 The presence of TG2 has been shown to be critical in the expression of calcium promotor genes in mouse and human aortic VSM cells. The effects of the calcium promoting stimuli of inorganic phosphate (Pi) and bone morphogenetic protein-2 (BMP-2) were studied on the function and phenotype of wild-type (WT) and TG2 knockout mouse cells. In the WT cells, Pi and BMP-2 caused upregulation of calcification promoter genes and formation of calcified nodules. The TG2 knockout cells changed from a contractile to a non-calcifying phenotype, and showed upregulation of calcification inhibitor genes.40 While this role for TG2 has been convincingly shown in cultured cells, it remains to be established in vivo,41 and how this can translate to modification of arterial stiffness.
Neurogenic mechanisms and arterial stiffness
Stiffness of the arterial wall is determined principally by the load-bearing structural components of elastin and collagen and the tone of the smooth muscle cells.42 Thus, signals that modulate smooth muscle tone can provide a means of regulation of arterial stiffness and the ensuing arterial haemodynamics. Heart rate has also been shown to affect PWV.43,44 The interrelationship between the distribution of wall tension between the passive wall components of elastin and collagen and the active component of smooth muscle is complex45 and depends on the relative contribution of the contracted smooth muscle to the overall wall stiffness. Smooth muscle content is of the order of 30% in the aorta but up to 75% in peripheral muscular arteries.42
While studies on neurological and hormonal control mechanisms in the circulation have focused essentially on resistance arteries,46 there have been investigations addressing the neurogenic influence on large arteries through the effect of smooth muscle tone.47 These have been varied and have produced inconsistent results in terms of quantifying the intrinsic neurogenic effect on the smooth muscle as separate from the passive mechanical stretch effect due to concomitant pressure changes. Generally, the overwhelming evidence suggests that stimulation of smooth muscle in elastic arteries can cause change in the wall mechanics, and so affect properties of compliance and wave transmission.1 However, the extent of the effect is not established. Studies simulating the neurogenic effect by administration of neurotransmitter substances have demonstrated increased aortic PWV in anaesthetized dogs48 and wall stiffness changes measured by pressure/diameter relations in conscious dogs33 and vagotomised cats.49 Studies in rats have also been confined to measurement of specific sites (carotid and femoral arteries) and have not addressed the effect on the aortic trunk50,51 in terms of functional stiffness determining pulse pressure. Recent studies in humans have shown an independent association between carotid-femoral PWV and muscle sympathetic nerve activity.52
Distribution of adrenergic nerve endings has been shown to be variable along the aorta.53 The chronic absence of sympathetic innervation has also been shown to produce a marked increase in collagen synthesis in the aortic wall.54
We have shown in previous studies in the sheep iliac artery that large artery stiffness can be modulated by local factors related to NO affecting endothelial function.55,56 We have also shown that this can be applied to the human iliac artery.57 The implications of this is that pharmacological therapies that target smooth muscle tone affecting wall stiffness of large arteries, which in turn affect the pulsatile haemodynamics of arterial function, can be used to reduce pulse pressure through effects of wall compliance and wave reflection. It is expected that similar mechanisms could be applied through the neurogenic effect on smooth muscle tone. Indeed, our recent pilot studies in changes in nocturnal blood pressure have shown a paradoxical change in pulse transit time of large conduit arteries, implicating possible active neurogenic mechanisms overriding passive pressure dependency of arterial stiffness.58
Conclusions
Developments in the biophysical aspects of arterial haemodynamics will lead to improved quantification of functional parameters based on established physical principles as was gained from early interaction of physiologists, mathematician and engineers, as exemplified by the classic work of Donald McDonald.1,10–12,48 Given the prominence of arterial stiffness as a fundamental property of pulsatile blood pressure and flow, wave propagation phenomena and its emergence as a powerful factor in cardiovascular risk,3,6,7,9,59,60 it is important to elucidate the underlying biological mechanisms that lead to medial degeneration of arterial properties that translate to functional correlates such as increased arterial systolic pressure. As in the early advances made with interdisplinary interaction that lead to coherent biophysical explanations of pulsatile blood flow in arteries, the improved understanding of the cellular, molecular and neurogenic mechanisms will come from a similar interaction of investigators from the broad field of physical sciences and molecular biology. Data to date suggest the existence of signaling pathways that form part of positive feedback mechanisms that can amplify the effects on arterial stiffness. This systems approach has the potential to lend itself to uncovering integrated mechanisms and parameters that can be associated with the merging fields of databases on genomics and proteomics. One such integrated parameter might well prove to be arterial wave velocity.
References
Cite this article
TY - JOUR AU - Alberto Avolio AU - Mark Butlin AU - Ying-Yi Liu AU - Kayla Viegas AU - Bhargava Avadhanam AU - George Lindesay PY - 2011 DA - 2011/11/06 TI - REGULATION OF ARTERIAL STIFFNESS: CELLULAR, MOLECULAR AND NEUROGENIC MECHANISMS☆ JO - Artery Research SP - 122 EP - 127 VL - 5 IS - 4 SN - 1876-4401 UR - https://doi.org/10.1016/j.artres.2011.10.002 DO - 10.1016/j.artres.2011.10.002 ID - Avolio2011 ER -