
Large arteries and angiotensin II blockade in hypertension
- DOI
- 10.1016/j.artres.2010.07.001How to use a DOI?
- Keywords
- Hypertension; Large arteries; Angiotensin
- Abstract
Renin-angiotensin blockade has reduced cardiovascular morbidity and mortality in hypertensive subjects. Experimental studies have shown that this blockade acts primarily on vascular tone, causing quasi simultaneously a decrease in vascular resistance, an increase in large artery compliance and a change in endothelial function independent of blood pressure changes. In hypertensive models, the reduction of arterial hypertensive hypertrophy is proportional to blood pressure reduction while the reduction of collagen is responsible for the elasticity improvement and only dependent on angiotensin II. Molecular biology shows that, under the control of MAP-kinase, the major substratum of the action is the biochemical complex between the integrin alpha5 beta1 and its ligand fibronectin, both highly sensitive to mechanical pulsatility and effects on collagen fibers. Following the Reason study, numerous therapeutic trials in hypertension have shown not only the antihypertensive effect of this complex but also its impact on pulsatility and finally on central systolic and pulse pressure.
- Copyright
- © 2010 Association for Research into Arterial Structure and Physiology. Published by Elsevier B.V. All rights reserved.
- Open Access
- This is an open access article distributed under the CC BY-NC license.
Introduction
Whereas blood flow is largely preserved in human and animal hypertension, arterial stiffness, evaluated from arterial distensibility, is significantly increased in the totality of the arterial tree.1–4 The increased arterial stiffness appears largely to be due to the elevated level of blood pressure as such, but there is increasing evidence that alterations of the arterial wall associated with hypertension also contribute to the changes in the viscoelastic arterial properties.4
To elucidate this point, arterial compliance and distensibility should be compared at the same pressure in the normotensive and the hypertensive populations (isobaric compliance and distensibility). In animal models of hypertension, studies of large arterial strips and rings indicate that isobaric compliance and distensibility are decreased, particularly in fully relaxed vessels.2,4 In human hypertension, for the same operational mean arterial pressure in the entire arterial tree, compliance and distensibility are decreased significantly in central arteries (aorta, carotid artery) but remain within the normal range in peripheral arteries (radial artery).2,4 On the other hand, direct histomorphometric studies of hypertensive large conduit arteries indicate that there are consistent structural alterations in proportion to the level of blood pressure. Such changes are also present in the coronary arteries independently of the occurrence of atherosclerosis.5,6 Considering that the clinical complications of hypertension are rather related to larger than smaller arteries, attention has been turned towards the possibility of, not only lowering blood pressure in hypertension but also reversing arterial structural and functional changes following treatment of hypertension. This question has been raised, in the frame work of hypertension, mainly by the use of blockade of the renin-angiotensin system.2,3
Experimental studies
Whereas, in most studies of the literature, the stiffness of the arterial wall in hypertensive animals has been evaluated on in vitro strips or rings of vascular tissue,2–4 investigations have been also carried out in living dogs and rats.7–9 In vivo studies using Doppler measurements of aortic wall movement have the advantage of giving dynamic values, but these values remain limited to the operational range of blood pressure of the corresponding animals. Furthermore, dynamic compliance so evaluated is a complex variable which is influenced not only by the changes in smooth muscle tone and in the structure of the arterial wall, but also by the frequency dependence of the elastic arterial wall and by the viscosity of the vessel. For pharmacological studies, it is important to minimize the two latter factors in order to evaluate predominantly the drug-induced changes in arterial structure and smooth muscle tone. In order to evaluate compliance, in situ preparations using 18–20 mm of non exposed common carotid artery have been prepared to establish the compliance-pressure relationship over a wide range of transmural pressure from 0 to 200 mmHg. Because the arterial tissue is heterogeneous8,10,11 and involves both smooth muscle and extracellular matrix, the compliance-pressure relationship was found curvilinear (Fig. 1). This indicates that (1) within the lower transmural pressure ranges, in which tension is borne by elastin and arterial smooth muscle tone, compliance increases markedly with increasing pressure, and (2) within the higher pressure ranges, tension is rather borne by collagen fibbers, causing compliance to decrease with pressure.
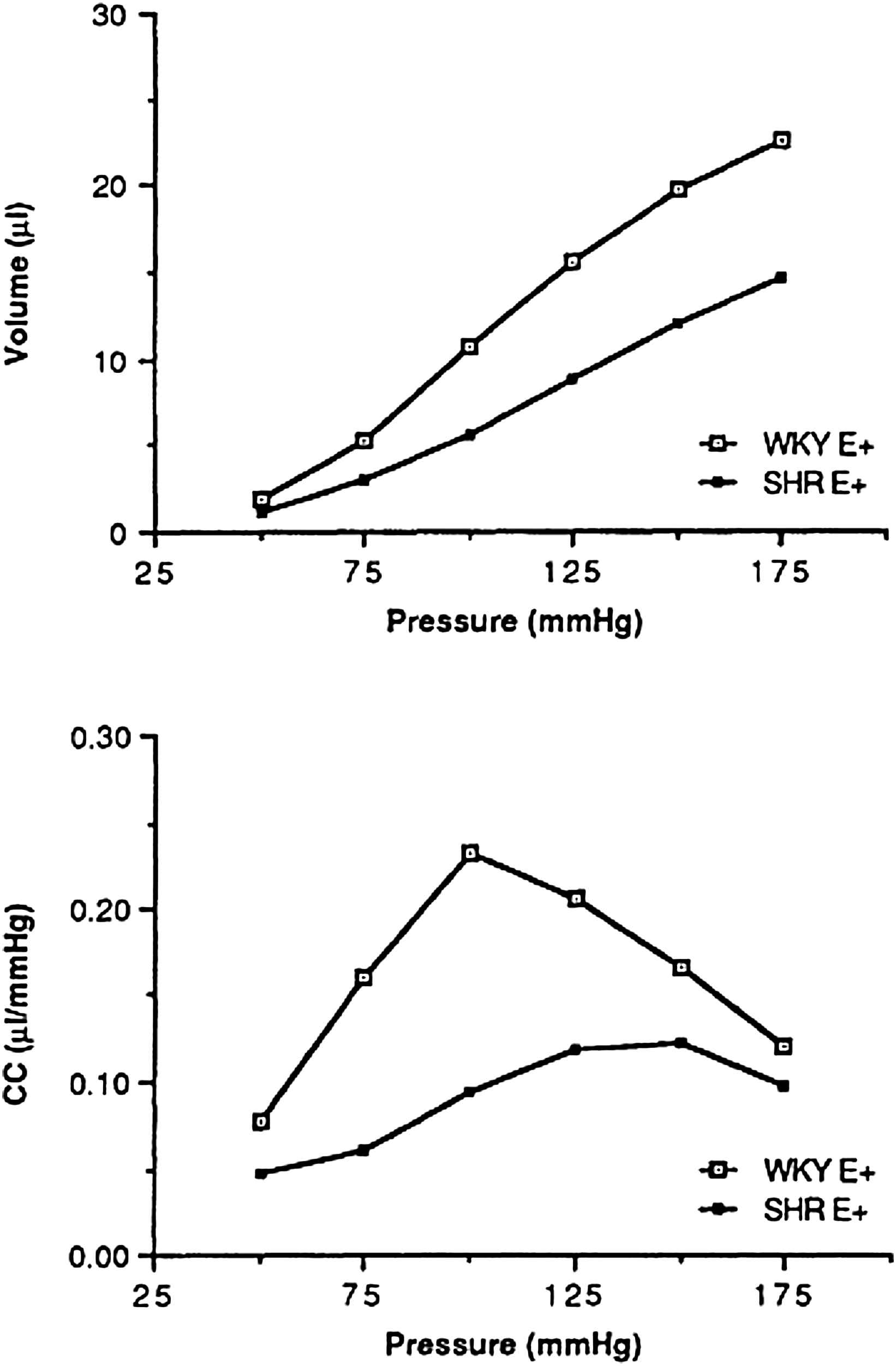
Top: Mean values of the carotid volume-pressure relation measured, with intact endothelium, in normotensive (WKY) and spontaneously hypertensive rats (SHR). Bottom: Compliance of the isolated carotid artery (CC) vs. pressure calculated as the slope of the volume-pressure curves (ΔV/ΔP) in WKY and SHR (mean ± SD).10
Studies on in situ carotid preparations show that converting enzyme inhibition increases compliance independently of transmural pressure, when given either locally10,12 or following acute oral administration. The compliance enhancement is obtained within the lower pressure ranges and disappears in the absence of endothelium, indicating that some of the effects of ACE inhibitors on viscoelastic properties of the arterial wall may depend on endothelial function.10 The same result is obtained using saralasin of bradykinin, instead of the converting enzyme inhibitor itself, but most studies suggest that the compliance increase is played mainly by Ang II blockade.13,14 The increase is noted even when the converting enzyme inhibitor given orally does not induce a significant reduction in systemic blood pressure,10,12 suggesting that a decrease in arterial smooth muscle tone alone may be associated with an improvement in the viscoelastic properties of the arterial wall.
In order to evaluate the role of structural factors on compliance modifications, the effects of chronic ACE inhibition on the function and the histomorphometric composition of the carotid artery and the aorta were studied in two-kidney-one-clip Goldblatt hypertensive rats, also in SHRs, and compared with age-matched normotensive rats.8,10,11 At the end of the treatment period, hemodynamic patterns were recorded and passive mechanical properties of the isolated carotid artery were measured in situ. Morphological parameters of the thoracic aortic wall, including media thickness, nucleus density, elastin and collagen contents, were measured using an automated system. In the hypertensive models, hypertension was associated with a stiffer arterial wall, as demonstrated by an increase in characteristic impedance of the aorta, a decrease in systemic arterial compliance, and a decrease in carotid compliance. Chronic ACE inhibition normalized blood pressure and reversed all these in vivo markers of vascular stiffness. The functional changes were obtained in parallel with structural modifications of the arterial wall. The increased aortic media thickness observed in hypertension was reversed. This reduction of the aortic media thickness was mainly related to a reduction of smooth muscle cell hypertrophy as assessed by the significant increase in nucleus density. There was no or only a slight decrease in the aortic collagen content. The differential effects on collagen could be related to differences in experimental models, and/or to the dose of ACE inhibition, and/or more likely to the duration of treatment (12 weeks in SHRs vs. 4 weeks in two-kidney-one-clip rats).
Because Ang II is known to stimulate collagen synthesis in cell cultures,15 the problem of aortic collagen content was then addressed using a preventive (and not a therapeutic) protocol.16,17 Four groups of 4-week-old SHRs were treated during 4 months with the angiotensin-converting enzyme inhibitor quinapril at 1 mg/kg/day (Q1) or 10 mg/kg/day (Q10), hydralazine at 15 mg/kg/day, or placebo.16 At the end of treatment, mean blood pressure was significantly lower in the Q10 and hydralazine groups (136 ± 16 and 149 ± 11 mmHg) compared with the placebo group (190 ± 23 mmHg). In the Q1group there was only a slight reduction in mean blood pressure with values lower than the placebo group but significantly higher that Q10 and hydralazine groups. The decrease in aortic medial cross-sectional area largely paralleled that of the blood pressure changes. In contrast, in the Q10 and the Q1groups, the collagen content of the aortic media was significantly lower than in the two other groups and no difference was observed between, Q1 and Q10 (Table 1). Aortic ACE activity was inhibited by approximately 60% in both groups treated with the ACE inhibitor, whereas plasma ACE activity was reduced only in the Q10 rats. These results show that whereas both hydralazine and quinapril prevented the development of aortic hypertrophy only in a pressure-dependent manner, the prevention of aortic collagen was observed only after ACE inhibition. This latter effect of ACE inhibition was not related to the blood pressure reduction but was associated with reduction of aortic (and not plasma)-converting enzyme. In a similar protocol using, in addition to quinapril, an angiotensin type I receptor antagonist (CI 996) and the bradykinin antagonist HOE 140, the results indicated that the prevention of aortic collagen accumulation was not due to bradykinin preservation but exclusively to the blockade of Ang II type 1 receptors.17
| Internal radius, μm | MCSA, μm2·103 | Collagen density, % | Elastin density, % | |
|---|---|---|---|---|
| PL | 772 ± 22 | 697 ± 21 | 20.5 ± 1.1 | 27.3 ± 1.3 |
| Q | 777 ± 27 | 611 ± 33a | 16.2 ± 1.1b | 21.7 ± 0.9b |
| QH | 756 ± 21 | 657 ± 30 | 15.2 ± 0.7b | 24.7 ± 0.8a,d |
| CI | 778 ± 20 | 670 ± 55 | 16.5 ± 1.0b | 21.7 ± 1.0b |
| WKY | 684 ± 9c | 526 ± 18b,e,f | 17.5 ± 0.6b | 23.7 ± 0.8a |
PL indicates placebo-treated SHRs; Q, quinapril-treated SHRs; QH, quinapril plus HOE 140–treated SHRs; CI, CI 996-treated SHRs; SHRs, spontaneously hypertensive rats; and WKY, Wistar-Kyoto rats treated with placebo. Values are mean ± SEM.
P < 0.05 vs placebo-treated SHRs.
P < 0.001 vs placebo-treated SHRs.
P < 0.01 vs others.
P < 0.05 vs Q and CI.
P < 0.001 vs QH and CI.
P < 0.05 vs Q.
Values of thoracic aorta internal radius, mean cross-sectional area (MCSA) and elastin and collagen densities in the different groups of rats.17
Taken together, all these findings indicate that Angiotensine II blockade act on both structure and function of both small and large hypertensive arteries, involving more specifically endothelial function, collagen accumulation and buffering function of conduit arteries.
Clinical studies
In recent years, non-invasive investigation of large arteries has been performed in humans using echo-Doppler techniques. The study of such arteries has some major methodological advantages when compared to small arteries. Firstly, operational blood pressure can be determined at the exact site where the arterial vessel is studied. Secondly, the viscoelastic properties of the arterial wall may be evaluated in vivo, under conditions of operating flow, smooth muscle tone and endothelial function. Thirdly, in hypertensive humans, determinations of arterial thickness may also be performed in vivo,5,6 allowing to calculate circumferential wall stress and elastic modulus. New echo-Doppler techniques have been developed in the recent years, allowing to evaluate the structure and function of conduit arteries with a high degree of reproducibility. In addition, new echo-tracking techniques of high resolution were developed in man to measure the inner diameter and the thickness of straight superficial arteries, such as the radial, the brachial and the common carotid arteries.7,18–20 Since, in clinical situations, it is not possible to evaluate the range of the whole pressure–volume relationship (i.e. from 0 to 200 mmHg) it has been necessary to develop and validate methods for the simultaneous determination of local and systemic dynamic compliance and distensibility using the determination of pulsatile changes of pressure and diameter.7,18,20,21 More recently, it has been possible to measure adequately carotid and radial artery wall thickness in humans and to show that both parameters are on average significantly increased in hypertensive patients.5,6 Under these conditions, the structural abnormalities of large or medium-sized arteries have been determined non-invasively in hypertensive patients, and this has been done before and after drug treatments.
Arterial diameters
In healthy volunteers on an unrestricted sodium diet, increasing doses of the converting enzyme inhibitor perindopril were administered orally.22 At low doses ACE inhibition induced preferential dilatation of arterioles. At higher doses increases in brachial arterial diameter were also obtained. Since plasma-converting enzyme activity was blocked even at the lower doses, diameter enlargement of the larger arteries obtained with higher doses of ACE inhibitor was suggested to be rather related to inhibition of tissue-converting enzyme.
In patients with sustained essential hypertension, the oral administration of captopril caused a significant acute increase in diameter of the brachial and, to a much smaller extent, of the carotid arteries.23 As in normotensive subjects, in hypertensive the increase in the diameter of the brachial artery with intravenous perindopril was obtained with higher doses than those needed for forearm arteriolar dilatation.3,4 With chronic oral administration of this ACE inhibitor, the increase in diameter was maintained during treatment periods of 3–12 months in hypertensive subjects.24,25 Since the above findings were obtained in the presence of a significant fall in blood pressure, it seems clear that the dilating effect of ACE inhibition overcame any pressure-related tendency for the arterial diameter to decrease. Depending on the balance between the antihypertensive or the dilating properties various degrees of diameter change have been obtained with different converting enzyme inhibitors.18,24–27 Finally, similar to the results obtained by Caputo et al.14 from animal experiments, clinical studies strongly suggest that in normotensive and hypertensive subjects, converting enzyme inhibition causes a dilatation of peripheral muscular arteries, independent of blood pressure changes.28
In humans, the pharmacological effects of converting enzyme inhibition on the arteriolar wall are considered to be mainly due to the blockade of the renin-angiotensin system. Injections of Ang I ad Ang II, in the presence or absence of various converting enzyme inhibitors, into the brachial artery indicate, the role of the renin-angiotensin system in regulating the forearm tone of small arteries.28 Other factors may also be involved, such as changes in endothelial function with local increases in bradykinin and prostaglandins, and attenuation of sympathetic activity.29–31 In man, it is possible that each of these factors contribute to large artery dilatation following converting enzyme inhibition. Several of these factors act by influencing endothelium-dependent vasodilation. Indeed, vascular endothelial cells are one of the specific sites of action for bradykinin and converting enzyme,30,32 and might be involved in producing an increase in brachial artery diameter following converting enzyme inhibition.
In hypertensive subjects, the increase in brachial artery diameter following ACE inhibition is associated with an increase in blood flow velocity.24,25 This might also contribute to the increase in arterial diameter via the mechanism of high flow dilatation which in turn is dependent upon the status of endothelium. Studies in dogs have shown that both epicardial coronary and femoral arteries dilate in response to an increase in blood flow.33 Any factor that increased flow, such as release of a transient arterial occlusion, causes dilatation of large arteries.34,35 If the increase in flow is prevented by a limiting stenosis, however, no dilatation of the large vessels occurs.34–37 Removal of the endothelium in isolated perfused canine coronary of femoral arteries in situ also abolishes flow-dependent dilatation. In a long-term study with perindopril in hypertensive subjects,24 the role of flow-dependent dilatation on the brachial artery was evaluated by studying the hemodynamic effects of wrist occlusion at a supra systolic blood pressure level. This manoeuvre caused a consistent reduction in brachial artery diameter and blood flow velocity during administration of either placebo or ACE inhibition. Although the brachial artery diameter decreased with wrist occlusion during active treatment, it remained greater than during placebo administration. These findings support the hypothesis that the flow-dependent dilatation and changes in endothelial function account in part for the increase in brachial artery diameter, but also suggest that flow-dependent vasodilatation is not the only mechanisms of action leading to arterial dilatation following ACE inhibition.
Arterial stiffness
In addition to diameter changes, long-term administration of converting enzyme inhibitors in patients with essential hypertension causes a significant increase in arterial compliance, which was observed at the site of the brachial, radial, femoral and carotid arteries.18,24,27,29 Several mechanisms may contribute to the increase in compliance. A reduction in blood pressure per se lessens the stretch of the arterial wall, thereby favouring the increase in compliance.1–4 During converting enzyme inhibition, however, the increase in diameter tends to maintain tangential tension despite the reduction in blood pressure.24 On the other hand, the direct or indirect effects of converting enzyme inhibition on arterial smooth muscle tone might have favor arterial smooth muscle relaxation with a resulting increase in compliance, as it has been observed in animal studies.8,38 The last possibility is the reversion of arterial structural changes following long-term treatment with converting enzyme inhibitors.
A study by Kool et al.18 in subjects with essential hypertension supports the idea that converting enzyme inhibition increases compliance independently of the level of the blood pressure. This study showed that for a similar antihypertensive efficacy, ACE inhibition improved carotid and femoral compliance whereas hydrochlorothiazide + amiloride had no effect. A similar finding was observed by Barenbrock et al..37 In a randomized double-blind study, the effect of lisinopril and metoprolol on arterial distensibility was studied in patients with hypertension and end-stage renal disease. After a placebo run-in period, the patients were randomly treated with metoprolol (50, 100 or 200 mg) or lisinopril (5, 10 or 20 mg) for 10 weeks of therapy. In both groups, a similar antihypertensive effect was observed. Neither ACE inhibition nor β-blockade influenced the end-diastolic diameter of the common carotid artery after 6 and 10 weeks of treatment. During ACE inhibition, a significant increase of the percent change in diameter and distensibility (compared with the metoprolol group) was observed. An increase in distensibility during ACE inhibition was further confirmed by others.18,39,40
Recently, Girerd et al.41 studied a large group of elderly patients with isolated systolic hypertension which were randomized in a double-blind parallel study, receiving either perindopril (2–8 mg) or hydrochlorothiazide + amiloride (hydrochlorothiazide 12.5–50 mg) during 9 months. If SBP remained above 160 mmHg after 5 months, atenolol or chlorothalidone was added. In both groups, there was a similar decreased in blood pressure. The ACE inhibitor and the diuretic compound significantly decreased the thickness of the radial artery and the cross-sectional area of the intima media. Such results demonstrated that the reversion of structural arterial changes can be obtained in old subjects with essential hypertension. However, in such old subjects with systolic hypertension, the reversibility of structural changes in large conduit arteries seemed to be influenced rather by the pressure-induced than by the drug-induced effect of treatment on the arterial wall. This finding, which differs from that of Kool et al.,18 indicates that different arterial alterations may occur in younger and older hypertensive subjects following drug treatment.
Finally, in these recent years, these apparent contradictions have been clarified on the basis on two news approaches. Fist, the understanding of wave reflections permitted us to better understand the complex relationship between blood pressure and the transduction of the arterial wall. Transduction mechanisms and molecular biology clarify now novel aspects of cardiovascular pharmacology in hypertension.
Blood pressure and wave reflections
Based on pulsatile arterial hemodynamics,1 it is widely accepted that the aortic blood pressure curve should be considered as the mathematical summation of an incident pressure wave, coming from the heart through ventricular ejection, and a reflected wave depending on a given site characterized by three independent parameters: the value of reflection coefficients, the degree of arterial stiffening and also the distance between the heart and the site of reflecting points. In normal conditions, the complex interaction between these parameters is responsible for the physiological amplification of the pressure pulse from central to peripheral arteries, whereas the mean arterial pressure remains almost constant along the arterial tree. In pathological conditions, as observed in aged people, particularly those with hypertension, both increased arterial stiffening and reflection sites closer the heart (i.e. reflection sites at the level of arteriolar or arterial bifurcations) cause the backward pressure wave to return sooner (i.e. during the systolic component of the aortic blood pressure curve), resulting in a greater summation between the forward and backward waves and consequently a higher aortic PP and systolic peak.3,4 Such changes are responsible in old subjects with hypertension for a peculiar hemodynamic pattern involving a disproportionate increase in SBP over DBP within the arterial tree and a loss in pulse pressure amplification. We refer to recent reviews3,4 for a detailed discussion of these basic hemodynamic considerations in hypertension. The point to consider here is that converting enzyme inhibitors are able not only to decrease mean blood pressure but also, through their effect on the conduit arteries, to decrease brachial pulse pressure39,42,43 and, even more selectively, carotid and aortic PP. We have mentioned earlier that converting enzyme inhibitors may increase arterial diameter and decrease arterial stiffness independently of changes in mean arterial pressure. On the other hand, converting enzyme inhibitors have been shown to act selectively on the backward aortic pressure wave, causing the reflected wave to return during the diastolic(and not the systolic) component of the blood pressure curve, thus reducing aortic pulse pressure and systolic peak much more than brachial pulse and SBP. Thus, the decrease in arterial stiffness and the change in the pattern of pulse wave reflections greatly contribute to a decrease in blood pressure through arterial (and not exclusively arteriolar) effects. Both in animals and in humans, there is strong evidence to support this assumption, particularly following long-term treatment with ACE inhibition. Firstly, in spontaneously hypertensive rats and in patients with systolic hypertension and end-stage renal disease, converting enzyme inhibition restores the aortic PP amplification normally observed from central to peripheral arteries, causing a more substantial decrease in carotid than in brachial systolic and PP.42,43 Secondly, in subjects with essential hypertension converting enzyme inhibitors decrease brachial SBP more than β-blocking agents for the same decrease in brachial DBP.
The REASON study44 was the first clinical trial to investigate the long-term effects of drug treatment on central SBP and PP, arterial stiffness and wave reflections. It was conducted in middle-aged hypertensive subjects. Administration of the ACEI perindopril (Per), combined with a low dose of the diuretic indapamide (Ind), for 1-year was compared to the effect of the beta-blocking agent atenolol. For the same DBP and MAP reduction, Per-Ind lowered SBP and PP more than atenolol (Fig. 2). Moreover the reduction was significantly more pronounced centrally (carotid artery) than peripherally (brachial artery). While the 2 drug regimens lowered PWV equally, only Per-Ind reduced central PP and Aix.1–4,44 Under atenolol, central PP and Aix remained unchanged. Furthermore, Per-Ind decreased cardiac hypertrophy more than atenolol. This effect was attributed to the Aix decline, indicating that the reduction of cardiac mass was related to that of central wave reflections.7,21,45 Finally, under drug treatment, the selective lowering of SBP (but not DBP) was significantly predicted by the baseline PWV value. Thus, the REASON study emphasized the predictive values of PWV, and central SBP and PP in the mechanisms of BP reduction in hypertensive subjects.46
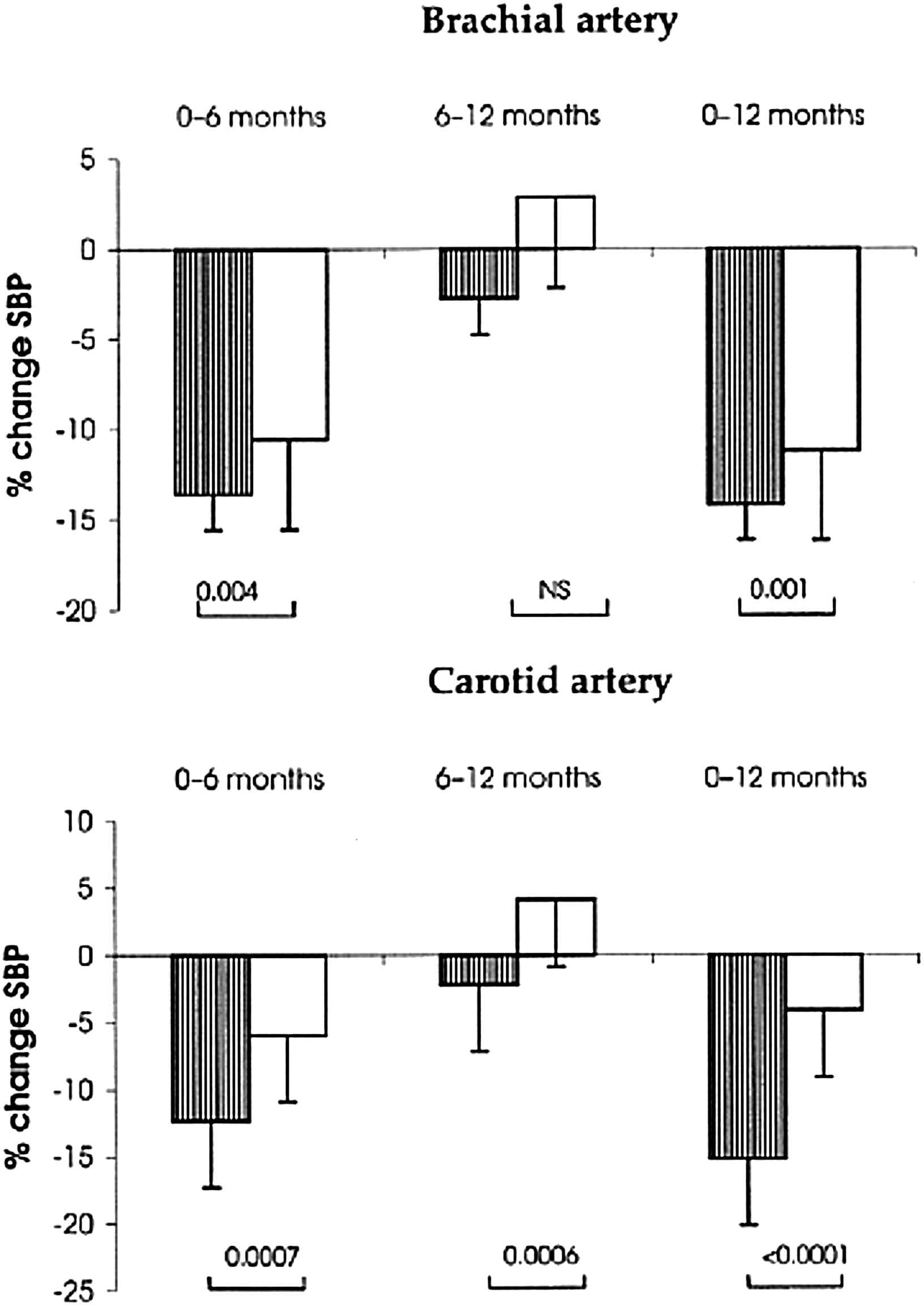
Hypertensive population with central measurements in the Reason study: adjusted changes (± SD) in systolic blood pressure (SBP) (%) during the evolution. With perindopril/indapamide (Per/Ind) (lined bars), SBP reduction (%) was more pronounced than for atenolol (open bars) at both the carotid and the brachial artery sites between 0–6 months and 0–12 months. Between 6 and 12 months, the SBP reduction continued under Per/Ind and tended to decline under atenolol. The p values are represented as Per/Ind versus atenolol.44
Transduction within the arterial wall, PP and ANG II blockade
Mechanical forces involve pulsatile and steady pressure components but should have to integrate some biological evidences such as: focal adhesion formations and integrin-specific signalling in VSM cells are essential for mechanotransduction of angiotensin blockade within the arterial wall. Furthermore, the endothelium is sensitive to mechanical solicitation by flow. Endothelial and NO signalling increases the arterial calibre during the vessel maturation and on arterial diameter during flow-induced vasodilatation. All these procedures should be at least partially to comment regarding angiotensin blockade.
Dense plaques, which are composed of cytoskeletal proteins linked to extracellular matrix (ECM) by integrin receptors, are major sites of anchorage between VSM and the ECM. They represent key elements for the distribution between mechanical and elastic materials. Integrins exist as αβ pairings. They interact with ECM components including fibronectin (ligand for α5β1 and αvβ3), vitronectin (ligand for αvβ3), and laminin (ligand for α6β1).47 Interactions of specific extracellular matrix (ECM) proteins with their integrin receptors play a central role in transmitting mechanical forces to VSM.48,49 A large number of these integrins are capable of forming complexes with fibronectin (Fn), a glycoprotein that plays an important role in the organization and assembly of the ECM.
When cyclic mechanical strain is applied to matrixes containing different adhesion proteins, Fn produces one of the largest mitogenic responses in rat VSM.49 In addition, expression of Fn and its α5β1 receptor are increased in SHR aorta. The increase of Fn may reflect an increased number of mechanical attachments between the ECM and collagen fibers within the media. From a mechanical point of view, an increased number of cell matrix attachments promote increased stiffness and mechanical strength.
These changes have been studied in situations involving normal or increased sodium intake in the presence of ANG II stimulation and blockade50–55 (Table 2). Aldosterone blockade was studied in parallel in the same conditions.53 With normal-sodium diet, angiotensin or aldosterone blockade reduced MAP and PP, and decreased collagen accumulation, Fn and its integrin receptor. On a high-sodium diet, MAP but not central PP, was reduced in association with collagen accumulation, increased Fn and an increased arterial stiffness.
| Normal-sodium diet | High-sodium diet | P Interaction (ANOVA) | |||
|---|---|---|---|---|---|
| Placebo (n = 14) | Valsartan (n = 15) | Placebo (n = 13) | Valsartan (n = 14) | ||
| Body weight, g | 412 ± 8 | 389 ± 10 | 382 ± 8 | 378 ± 11 | NS |
| Mean arterial pressure (mmHg) | 182 ± 6 | 144 ± 3* | 214 ± 6 | 180 ± 5* | NS |
| PP, mmHg (PP) | 60 ± 3 | 42 ± 3* | 69 ± 2 | 69 ± 3 | <0.002 |
| Heart rate, bpm | 349 ± 11 | 336 ± 10 | 347 ± 12 | 363 ± 11 | NS |
| Distensibility, mmHg−1·10−3 | 2.01 ± 0.16 | 4.43 ± 0.40* | 1.37 ± 0.11 | 1.61 ± 0.21 | <0.0001 |
| Einc, kPa | 1176 ± 121 | 539 ± 53* | 1659 ± 137 | 1679 ± 279 | 0.07 |
| Wall stress, kPa | 210 ± 9 | 168 ± 9* | 242 ± 11 | 198 ± 12 | NS |
Values are mean ± SEM.
P < 0.05 vs placebo for the same salt diet.
P < 0.05, HSD vs NSD for the same treatment.
Einc: incremental elastic modulus.
Effect of valsartan on body weight, blood pressure, and carotid artery parameters in SHR receiving a normal-sodium diet (NSD) or a high-sodium diet (HSD). Note that, under NSD, valsartan normalizes PP, not MAP. Under HSD, valsartan reduces MAP and does not modify PP.52
It is worth noting that the mechanical contribution of MAP and PP may vary markedly according to age or the vascular territory involved.56 Thus, the PP mechanotransduction may predominate in the heart rather than in the brain or kidney since only the former territory involves active periodic auto contractions. Furthermore, with age, the reflection coefficients are located much closer to target organs (i.e., heart, brain or kidney). This finding suggests that pulsatility and wave reflections may be transmitted to these organs in the presence of a defect in myogenic tone. This defect is commonly observed in subjects with type 2 diabetes mellitus, subjects with uninephrectomy, some obese patients, and older patients with systolic hypertension.57–59 Finally, many factors linked to mechanical stress, particularly pulsatility and wave reflections, may affect locally each particular organ, thereby modulating or interacting with local hormonal system.
In conclusion, the renin-angiotensin-aldosterone system has been considered primarily as a hormonal factor at the origin of hypertension and his treatment. With the development of antihypertensive therapy, the same system became an anti oxidadative and anti-inflammatory factor contributing to the reduction of morbidity and mortality. Nowadays, within the fields of the improvement of large arteries ANG II blockade acts mainly through its anti-fibrotic action and its development.
Acknowledgements
This study was performed with the help of INSERM (Institut de la Santé et de la Recherche Médicale) and GPH-CV (Groupe de Pharmacologie et d’Hémodynamique Cardiovasculaire), Paris. We thank Dr. Anne Safar for helpful and stimulating discussions.
References
Cite this article
TY - JOUR AU - Michel E. Safar PY - 2010 DA - 2010/09/16 TI - Large arteries and angiotensin II blockade in hypertension JO - Artery Research SP - 67 EP - 74 VL - 4 IS - 3 SN - 1876-4401 UR - https://doi.org/10.1016/j.artres.2010.07.001 DO - 10.1016/j.artres.2010.07.001 ID - Safar2010 ER -