
Loss of elastic fiber integrity compromises common carotid artery function: Implications for vascular aging
- DOI
- 10.1016/j.artres.2016.04.001How to use a DOI?
- Keywords
- Elastin; Fibrillin-1; Fibulin-5; Elastic energy storage; Distensibility; Pulse wave velocity
- Abstract
Competent elastic fibers endow central arteries with the compliance and resilience that are fundamental to their primary mechanical function in vertebrates. That is, by enabling elastic energy to be stored in the arterial wall during systole and then to be used to work on the blood during diastole, elastic fibers decrease ventricular workload and augment blood flow in pulsatile systems. Indeed, because elastic fibers are formed during development and stretched during somatic growth, their continual tendency to recoil contributes to the undulation of the stiffer collagen fibers, which facilitates further the overall compliance of the wall under physiologic pressures while allowing the collagen to limit over-distension during acute increases in blood pressure. In this paper, we use consistent methods of measurement and quantification to compare the biaxial material stiffness, structural stiffness, and energy storage capacity of murine common carotid arteries having graded degrees of elastic fiber integrity – normal, elastin-deficient, fibrillin-1 deficient, fibulin-5 null, and elastase-treated. The finding that the intrinsic material stiffness tends to be maintained nearly constant suggests that intramural cells seek to maintain a favorable micromechanical environment in which to function. Nevertheless, a loss of elastic energy storage capability due to the loss of elastic fiber integrity severely compromises the primary function of these central arteries.
- Copyright
- © 2016 Association for Research into Arterial Structure and Physiology. Published by Elsevier B.V. All rights reserved.
- Open Access
- This is an open access article distributed under the CC BY-NC license.
Introduction
The mechanical functionality, structural integrity, and adaptive capacity of central arteries result in large part from cellular regulation of the myriad structural constituents that constitute the wall, and their interactions. Notwithstanding the many contributors to the mechanics of the arterial wall, four are of primary importance: elastic fibers, fibrillar collagens, contractile smooth muscle, and proteoglycans/glycosaminoglycans.1,2 Of these four, elastic fibers are unique - they alone do not turnover during maturity and they are not effectively repaired or replaced during disease or injury. That is, functional elastic fibers are produced prior to adulthood and they have a half-life on the order of 25–70 years in both mice and humans under normal conditions.3–6 These fibers are thus uniquely susceptible to damage by mechanical fatigue in addition to normal degradation by proteolysis.7–9 Because elastic fibers cannot be effectively repaired or replaced, congenital and acquired losses of elastic fiber integrity have irreversible consequences on arterial mechanics and mechanobiology, and thus the physiology and pathophysiology. Genetically induced changes in, proteolytic degradation of, or mechanical damage to elastic fibers contribute, therefore, to central artery stiffening in aging and hypertension, which in turn promote diverse cardiovascular, renovascular, and neurovascular diseases.10–12 Indeed, loss of elastic fiber integrity also plays direct mechanical roles in atherosclerosis, aneurysms, and dissections.13–15
Elastic fibers in central arteries are organized into concentric fenestrated elastic sheets, or laminae, that form a three-dimensional network in the media. These fibers consist primarily of a core of elastin (90%) plus multiple elastin-associated glycoproteins.2,16 The glycoproteins fibulin-5 and fibrillin-1 are fundamental to the elastogenesis and long term mechanical and biological stability, respectively, of the composite fibers.17,18 Elastic fibers also associate with proteoglycans and minor collagens (e.g., collagen VIII), though specific contributions of these constituents to mechanical functionality is less clear.16 Nevertheless, loss of elastic fiber integrity has five primary consequences to the arterial wall: loss of resilience and thus a decrease in elastic recoil,19 decrease in collagen fiber undulation and thus diminished distensibility and extensibility,20 loss of cell–matrix interactions and thus altered smooth muscle cell phenotypic activity,17,21 increased intramural inflammation due to the chemotactic character of elastin degradation products,9 and possible premature entrance into the cell death cycle, or anoikis.22
Among the many important consequences of reduced elastic fiber integrity, we focused on the mechanical functionality of the common carotid artery (CCA) in the mouse. Specifically, we contrasted passive biaxial mechanical behaviors of carotids from control, elastin haploinsufficient (Eln+/−), fibrillin-1 deficient (Fbn1mgR/mgR), and fibulin-5 null (Fbln5−/−) mice with elastase-treated carotids from control mice. In this way, we assessed graded effects on wall structure and function due to elastic fibers ranging from normal to fully compromised. New biaxial data (Eln+/−) are presented and compared with previously published data (controls from,23 elastase-treated and Fbn1mgR/mgR from,20 and Fbln5−/− from 24) using new approaches for interpreting the data. By treating control and elastase-treated data as upper and lower bounds of elastic fiber functionality, respectively, we elucidated graded changes in carotid artery mechanics across the three genetic models. Importantly, total elastic energy displays a strong qualitative correlation with the severity of elastic fiber disarray.
Methods
Experimental methods
All animal protocols were approved by the Institutional Animal Care and Use Committee of Yale University. Briefly, mice were euthanized and CCAs were gently excised, cleaned of excess perivascular tissue, cannulated on custom drawn glass pipettes, and placed within a custom biaxial testing system in a Hanks buffered saline at 37 °C25; we have shown that this protocol yields a passive behavior.20 Following methods described previously,23 we performed cyclic pressure–diameter (P–d) tests at three different fixed axial stretches (the in vivo value and 5% above and below this value) and cyclic axial force–length (f–l) tests at four different fixed pressures (10, 60, 100, 140 mmHg). The latter tests are particularly important for estimating the preferred, or optimal, in vivo axial stretch26 and ensuring robust estimations of material parameters that define biaxial wall properties.23 All data (luminal pressure, outer diameter, axial force, and axial length, noting that axial stretch is defined as the ratio of the in vivo to the unloaded in vitro length) were collected using a custom LabView program and used for on-line feedback control of the tests and off-line analysis.
Material characterization
Each of the seven biaxial protocols (three P–d plus four f–l) consisted of two cycles of data. We extracted and combined the unloading portion of the final cycle for the seven data sets, each of which consisted of hundreds of data points. The unloading portion of a testing cycle provides information on the energy stored in the tissue that is yet available to work on the blood; that is, it does not include the small amount (typically 3–5%) of energy that dissipates during full pressure–diameter and axial force–length testing. The resulting combined data sets were analyzed via nonlinear regression to identify best-fit values of model parameters in a validated constitutive relation. Details on the constitutive formulation and regression method are given elsewhere.23,27 Briefly, we use a stored-energy function W containing 8 parameters
Physiological predictions
The primary loads acting on the CCA in vivo are an axial stretch-associated axial force and the transmural pressure. The former, which results primarily from somatic growth, is controlled locally by intramural cells and can change over time due to growth and remodeling.26 Multiple studies confirm that the associated value of in vivo axial stretch can be inferred via a “cross-over point” in in vitro f–l tests.31,32 The in vivo transmural pressure is influenced by systemic factors, notably cardiac output and peripheral vascular resistance, and by local effects of perivascular tissues. The latter are difficult to assess, hence transmural pressures are typically assumed to be slightly below measured luminal pressures. We extracted blood pressure data from the literature that were collected invasively under anesthesia, thus representing direct measurements of central blood pressure. Our selection criteria included the need to match mouse genotype and age with those animals that underwent biomechanical testing. Final values are listed in Table 1 and discussed below.
| Control | Eln+/− | Fbn1mgR/mgR | Fbln5−/− | Elastase | |
|---|---|---|---|---|---|
| n | 16 | 6 | 6 | 5 | 7 |
| Age (weeks) | 9.0 ± 0.4 | 8.0 ± 0 | 8.0 ± 0.2 | 21.3 ± 0.7* | 8.2 ± 0.3 |
| Body Mass (g) | 22.9 ± 0.9 | 25.2 ± 0.8 | 20.1 ± 2.3 | 33.0 ± 2.2* | 21.7 ± 0.9 |
| Systolic Blood Pressure (mm Hg) | |||||
| Systolic | 110a,b | 128a | 131c | 112b | 110a,b |
| Pulse | 35a,b | 48a | 43c | 48b | 35a,b |
| Unloaded Dimensions | |||||
| Outer Diameter (μm) | 394 ± 8 | 374 ± 11 | 399 ± 6 | 417 ± 13 | 693 ± 17* |
| Wall Thickness (μm) | 94 ± 4 | 81 ± 5 | 94 ± 5 | 72 ± 4* | 27 ± 1 |
| In-vitro Axial Length (mm) | 4.66 ± 0.21 | 5.91 ± 0.20 | 4.14 ± 0.43 | 5.42 ± 0.34 | 7.80 ± 0.5* |
| Systolic Dimensions | |||||
| Outer Diameter (μm) | 641 ± 9 | 592 ± 11 | 656 ± 16 | 554 ± 12* | 734 ± 20* |
| Wall Thickness (μm) | 26 ± 1 | 25 ± 2 | 28 ± 1 | 34 ± 2* | 24 ± 1 |
| In-vivo Axial Stretch | 1.72 ± 0.02 | 1.68 ± 0.03 | 1.61 ± 0.03* | 1.40 ± 0.02* | 1.06 ± 0.02* |
| Systolic Cauchy Stresses (kPa) | |||||
| Circumferential | 164.5 ± 4.6 | 190.4 ± 14.3 | 186.9 ± 5.6 | 109.2 ± 10.1* | 215.1 ± 17.5* |
| Axial | 190.4 ± 7.1 | 225.3 ± 20.4 | 164.8 ± 6.2 | 142.0 ± 16.4 | 176.0 ± 14.2 |
| Systolic Stiffness (MPa) | |||||
| Circumferential | 1.30 ± 0.09 | 1.88 ± 0.23 | 1.78 ± 0.10 | 1.07 ± 0.17 | 6.83 ± 1.32* |
| Axial | 2.75 ± 0.23 | 4.52 ± 0.46* | 2.45 ± 0.29 | 3.29 ± 0.37 | 3.97 ± 0.66 |
| Stored Energy (kPa) | |||||
| Systole | 48.6 ± 2.2 | 49.4 ± 4.7 | 36.4 ± 1.5* | 22.7 ± 2.2* | 7.8 ± 0.6* |
| Cardiac Cycle | 11.5 ± 1.1 | 12.0 ± 2.0 | 9.1 ± 0.9 | 7.6 ± 0.9 | 3.2 ± 0.8* |
Mean physiologic and mechanical data for common carotid arteries. Systolic and pulse pressures were extracted from aLe et al.,47 bLe et al.,53 and cMarque et al.40 We used data for C57BL/6J and Eln+/− mice, from47 and on mixed C57BL/6J × 129/SvEv, as controls, all at 60 days of age, which matches well the 8–9 weeks of age in most groups. Control pressures were averaged from the two studies. Fbln5−/− animals were significantly older than 60 days, but blood pressure was assumed to be similar at 21 weeks based on the observation of stable geometrical and mechanical properties between 8 and 14 weeks of age by Wan et al.49 Fbn1mgR/mgR pressures were measured by40 using a different anesthetic, catheterization method, and older mice. Control pressures were used to simulate the mechanics of elastase-treated carotids in vivo. All data are presented as mean ± standard error of the mean. An * denotes a significant difference with respect to control based on one-way ANOVA, with a post-hoc Bonferroni correction, and a p < 0.05 significance level.
Mutations of genes that code elastic fiber proteins affect blood pressure,33 but variations due to experimental conditions - including depth of anesthesia - are important as well. Thus, we performed a sensitivity analysis to understand how changes in pressure affect the biomechanical metrics of interest for the different groups studied. Specifically, we assessed different mechanical metrics of interest over transmural pressures from 50 to 150 mmHg, in increments of 10 mmHg, to cover a wide range of physiologic values.
Statistical analysis
All data are reported as mean ± the standard error of the mean (SEM). Comparisons between groups were made using a one-way ANOVA with post-hoc Bonferroni correction and a value of p < 0.05 considered significant. Significant difference from control is indicated in figures and tables, were appropriate.
Results
Table 1 provides morphometric, hemodynamic, geometric, and mechanical information for all 5 groups: control, Eln+/−, Fbn1mgR/mgR, Fbln5−/−, and elastase-treated. Figure 1 shows mean pressure–diameter behaviors at group-specific values of axial stretch, and axial force-axial stretch responses at a common pressure of 100 mmHg. Note the marked decrease in extensibility with the loss of elastic fiber integrity, as evidenced by decreasing values of mean in vivo axial stretch from 1.72 (control) to 1.68 (Eln+/−), 1.61 (Fbn1mgR/mgR), 1.40 (Fbln5−/−), and especially elastase-treated (1.06) vessels. Acute enzymatic removal of elastin also causes a marked dilatation and loss of distensibility, consistent with a vessel consisting primarily of collagen fibers having modest undulation.20 Control and elastase-treated CCAs thus served as upper and lower bound behaviors for the three genetically modified models. As it can be seen, the pressure–diameter behavior of Fbn1mgR/mgR carotids is most like that of the control while the behavior of the Eln+/− carotids is qualitatively similar, but shifted to the left due to smaller radii. Although similar at low pressures, the behavior of the Fbln5−/− group differs most from that of the control at physiologic pressures. These structural (P–d, f–l) responses are relevant to in vivo effects of the wall on the hemodynamics, but material (stress-stretch) behaviors reveal intrinsic properties.
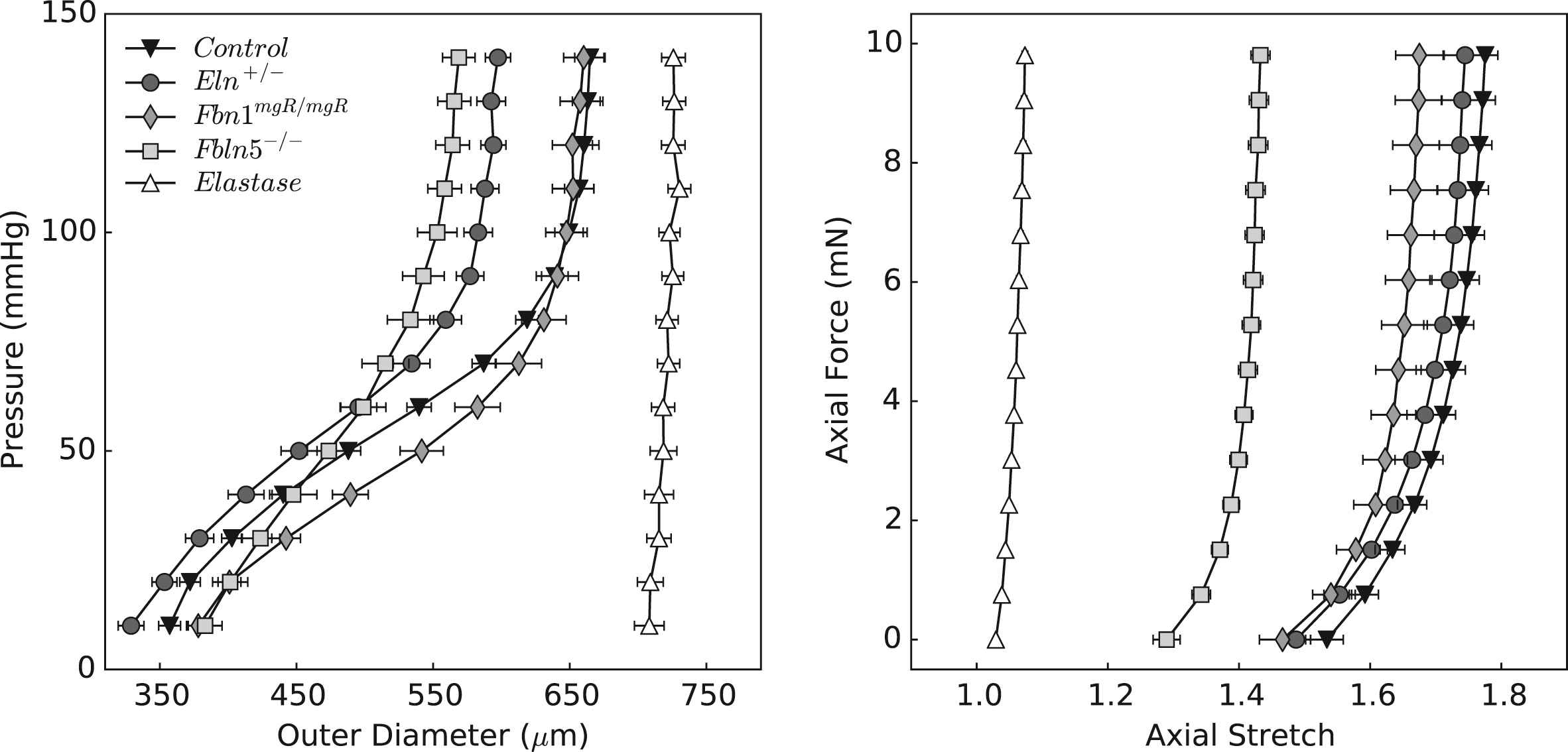
Structural behavior of common carotid arteries upon biaxial testing. Data are presented as mean ± standard error of the mean, with each curve representing the average of the respective experimental group. The left panel shows pressure–diameter responses during inflation tests at the estimated values of in vivo axial stretch; the right panel shows axial force-stretch responses at a common distending pressure of 100 mmHg. Note the considerable reproducibility achieved under computer-control.
Figure 2 shows the associated mean stress-stretch behaviors in circumferential and axial directions, while Table 2 lists associated values of the best-fit model parameters. Comparing pressure–diameter and circumferential stress-stretch responses reveals the utility of the latter in characterizing material behavior independent of geometry. The stress-stretch responses “fanned-out” and revealed more clearly the different degrees of distensibility, which tend to mirror losses of extensibility. In particular, the Fbln5−/− mutation yielded the most severe elastopathy of the genetically modified models (including the lowest value of in vivo axial stretch). Although we are tempted to interpret these results by stating that the Fbln5−/− carotid is the “stiffest”, it is better to observe that it is simply the least distensible and extensible of the three mutant models. Indeed, computations under in vivo loads reveal that circumferential stiffness is similar amongst controls and genetically modified models, though it is significantly higher in elastase-treated vessels. Axial stiffness is significantly higher in Eln+/− vessels and generally higher in the elastase-treated vessels. To avoid interpretative issues that arise because of pressure dependence, we calculated biaxial stiffness over a fixed range of blood pressures (50–150 mmHg). Figure 3 shows that stiffness increases nearly linearly with associated values of stress, as expected of a highly nonlinear (exponential) material behavior,34 with circumferential results very similar across all models except elastase-treated. Note that changes due to elastase treatment result from an acute insult, not a germline mutation, the latter of which allow possible compensatory remodeling throughout development. That neither circumferential nor axial material stiffness differed dramatically between the mutants and control, when computed at individual in vivo axial deformations, may reflect mechanobiologically favorable compensatory responses of CCAs during development in these mice.

Material properties of common carotid arteries with different degrees of elastic fiber integrity. By treating carotid arteries as thin-walled cylinders, one can calculate principal deformations and stresses from experimental data simply by imposing mechanical equilibrium (see Appendix). Cauchy stress-stretch responses are shown for inflation tests at the estimated values of in vivo axial stretch (circumferential properties, left panel), and for extension tests at 100 mmHg (axial properties, right panel). Note the different scales for circumferential and axial stretches. Propagation of experimental errors was calculated using the basic theory of error analysis,65 taking into account errors in the unloaded configuration and each of the loaded configurations.
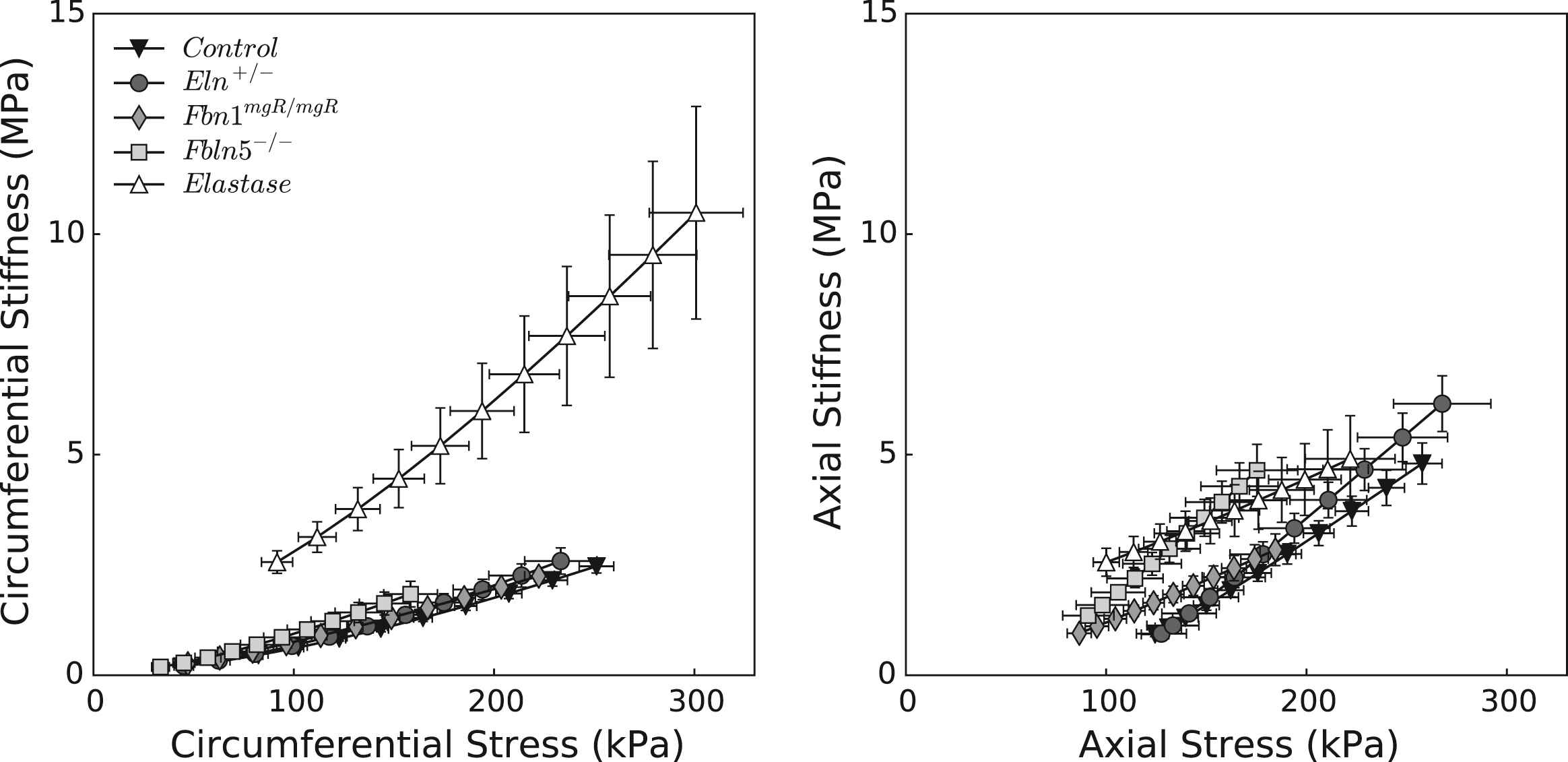
Material stiffness plotted versus Cauchy stress for the five groups of common carotid arteries. All three genetic models lie on the same circumferential stiffness–stress relationship as the control (left panel), while more heterogeneity is observed axially (right panel). Elastase-treated carotids display the most abnormal behavior, with significantly higher circumferential stiffness values at all pressures. This finding suggests that in vivo stiffness depends largely on the non-elastin components of the matrix, at least given the pressures considered.
| Elastic fibers | Axial collagen | Circumferential collagen | Diagonal collagen | RMSE | |||||
|---|---|---|---|---|---|---|---|---|---|
| c (kPa) |
|
|
|
|
|
|
α0 (°) | ||
| Control | 8.610 | 9.299 | 0.142 | 4.467 | 0.045 | 0.015 | 1.184 | 29.011 | 0.098 |
| Eln+/− | 11.753 | 9.645 | 0.176 | 5.104 | 0.088 | 9.46 × 10−4 | 2.156 | 30.922 | 0.095 |
| Fbn1mgR/mgR | 6.928 | 0.301 | 1.174 | 0.937 | 0.179 | 1.436 | 0.554 | 31.988 | 0.119 |
| Fbln5−/− | 21.233 | 2.171 | 2.581 | 4.551 | 0.833 | 0.015 | 6.505 | 26.282 | 0.079 |
| Elastase | 6.78 × 10−12 | 126.976 | 17.906 | 5.84 × 10−8 | 7.00 × 10−13 | 919.416 | 23.724 | 46.988 | 0.282 |
Recalling that the primary function of a large artery is to store elastic energy, Fig. 4 shows iso-energy contours for all 5 groups plus associated histological sections. By plotting energy as a function of biaxial stretch (circumferential along the ordinate and axial along the abscissa), one can easily visualize the degrees of distensibility, extensibility, and anisotropy. Again, control and elastase-treated vessels provide natural upper and lower bounds of elastic fiber integrity. Despite having smaller diameters, Eln+/− carotids exhibit an adaptive increase in medial lamellar units that results in near normal values of energy storage at systole (Table 1). Fbn1mgR/mgR carotids store less energy and exhibit a stronger anisotropy than controls or Eln+/− vessels, primarily due to the well-known reduction of in vivo axial stretch.20,29 Their anisotropic energy map is consistent with the hypothesis26 that an accelerated loss of elastic fibers in Marfan mouse models (confirmed by focal interruptions in the lamellar structure) is compensated by a reduced axial stretch, which helps to promote physiologic levels of distensibility, stress, and stiffness. Fbln5−/− carotids exhibit the most dramatic reduction in energy storage, that is, the most severe elastopathy of the three mutants. These vessels are distinguished by a more generalized elastic fiber fragmentation, particularly in the outer media, with clusters of nonfunctional (i.e., non-load bearing) elastin neighboring the main lamellar structures; this radial gradient in elastic matrix disorganization is similar to that previously reported in the thoracic aorta of these mice.35,36 It appears that the confined stored energy contours and modest values of energy storage at systole result from these underlying structural deficiencies, which prevent Fbln5−/− vessels from undergoing normal axial and circumferential deformations. Indeed, results from elastase-treated carotids show that a near total loss of elastin results in minimal axial and circumferential deformations and the lowest degree of elastic energy storage, both of which result in part from a near immediate engagement of adventitial collagen upon loading.
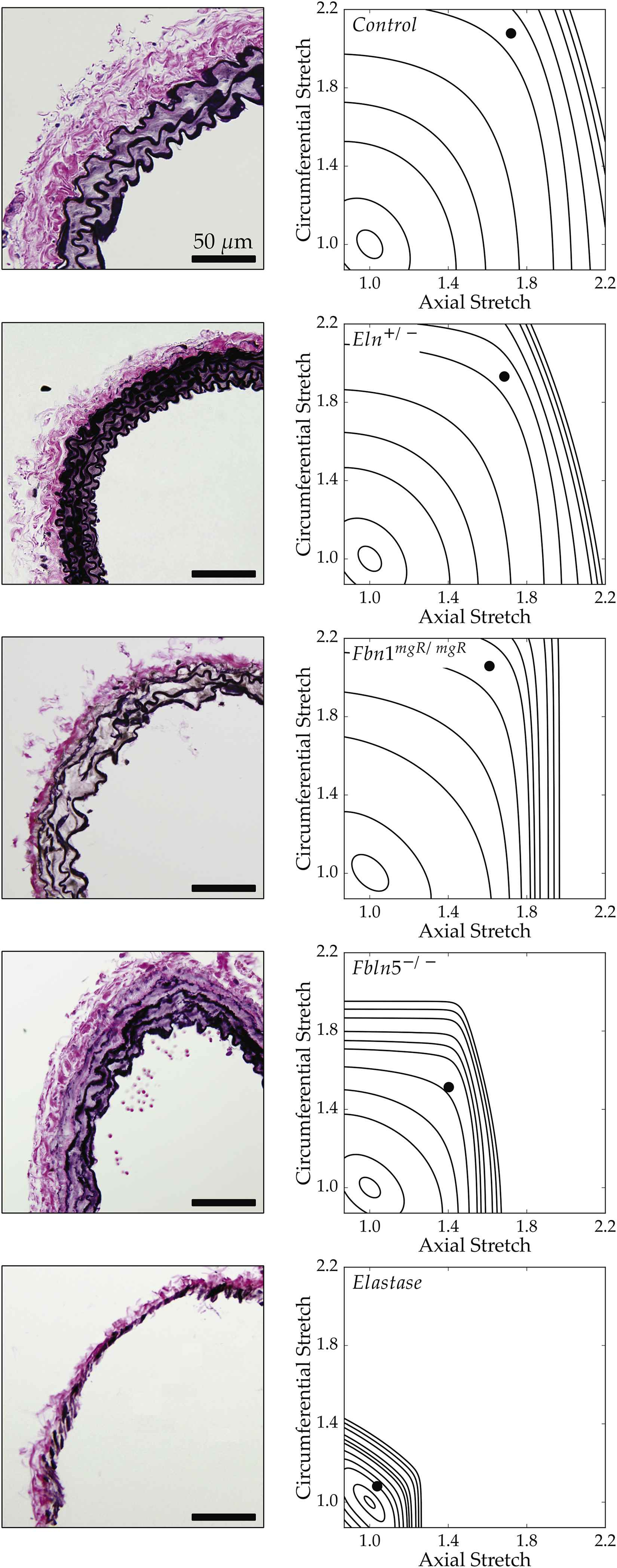
Bright-field microscopy images of Verhoeff Van Gieson (VVG) stained histological cross sections (left column) paired with stored energy contours calculated as a function of biaxial stretches (see Appendix). Filled circles represent in vivo energy values computed at group-specific values of in vivo axial stretch and systolic pressure relative to an unloaded configuration. This qualitative comparison shows that less energy is stored elastically for greater degrees of elastic fiber insult.
Figure 5 highlights these differences in biaxial deformations and energy storage across groups. Circumferential and especially axial stretches diminish with loss of elastic fiber integrity, with a pattern for the in vivo axial stretch that matches closely the loss of energy storage at systolic pressure. When calculated relative to the unloaded state, systolic energy storage in Fbn1mgR/mgR, Fbln5−/−, and elastase-treated carotids is, respectively, 74.9%, 46.7%, and 16.0% of normal; when calculated over a single cardiac cycle (see Appendix), cyclic energy storage for the same mutants is 79.1%, 66.1%, and 27.8% of normal. The latter reductions did not reach statistical significance in any of the genetic models, however. Nevertheless, these results highlight how loss of elastic fiber integrity primarily affects arterial extensibility, which in turn is a key determinant of total energy storage.
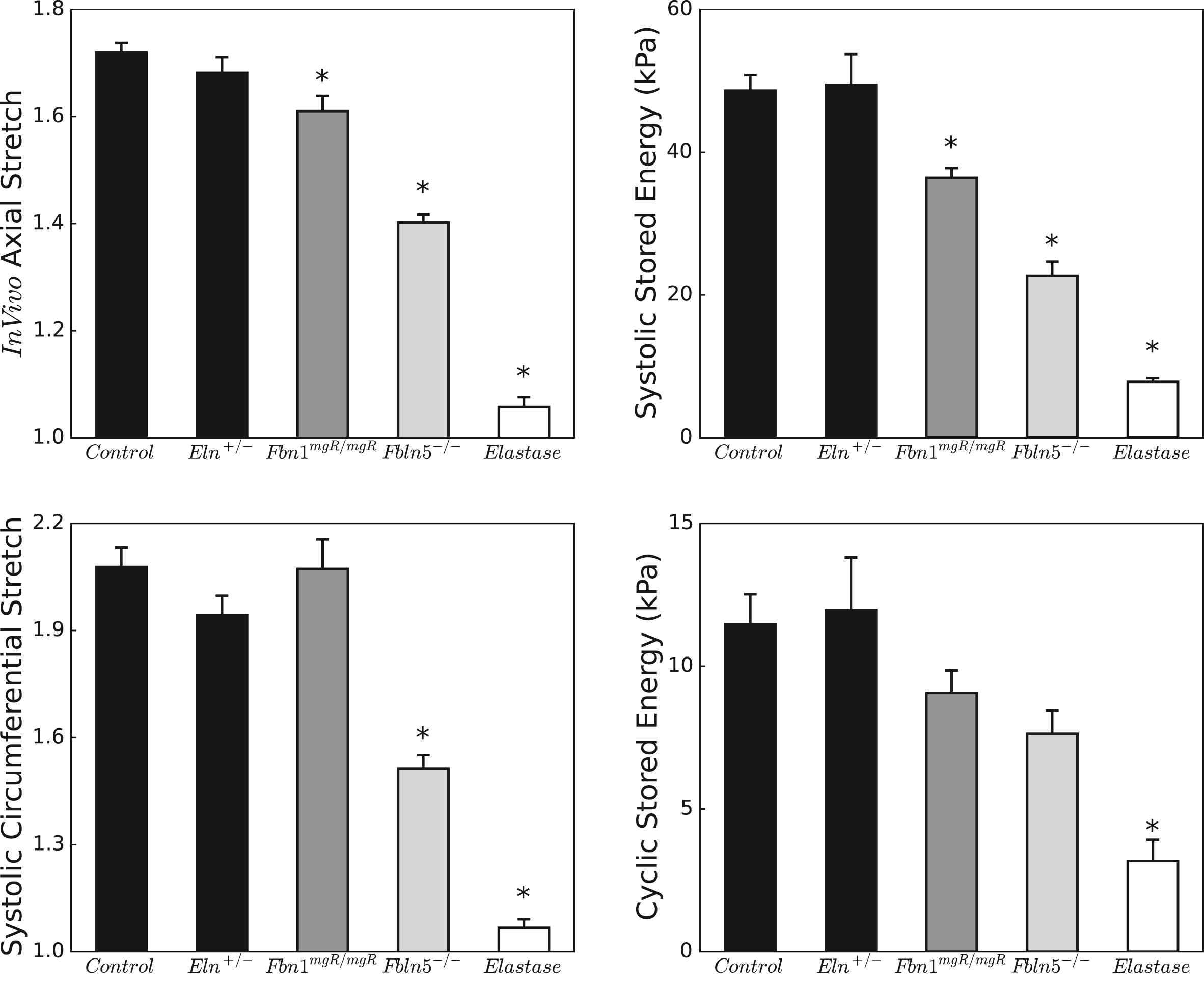
Biaxial stretches (left column) and stored energies (right column) under physiologic conditions. Note the different scales for the stretches, with axial stretches exhibiting a more limited range due to the well-known anisotropy of the arterial wall. Stored energies computed at individual systolic pressures are similar between control and Eln+/− carotids, but decrease progressively in Fbn1mgR/mgR, Fbln5−/−, and elastase-treated carotids, following a trend similar to the in vivo axial stretch. Relative energy storage (over a cardiac cycle) follows the same pattern as total energy storage but loses statistical significance with n = 5–7. The * indicates significant differences (p < 0.05) with respect to control. Corresponding numerical values can be found in Table 1.
Energy storage – like stress, material stiffness, and most other mechanical metrics – depends on the value of the distending pressure. Figure 6 shows that stored energy increases gradually with pressure, at group-specific fixed values of axial stretch, with all five models preserving their relative energy storage capability. Although energy storage is a functional-metric of intrinsic material behavior, it cannot be measured directly in vivo. More convenient metrics of structural behavior are often measured and compared. Figure 6 also shows values of local pulse wave velocity (PWV), estimated using the Moens–Korteweg equation (see Appendix), for all five groups as a function of pressure. With the exception of elastase-treated vessels, values of PWV are indistinguishable between control and mutant arteries; they simply vary nearly linearly with the distending pressure.
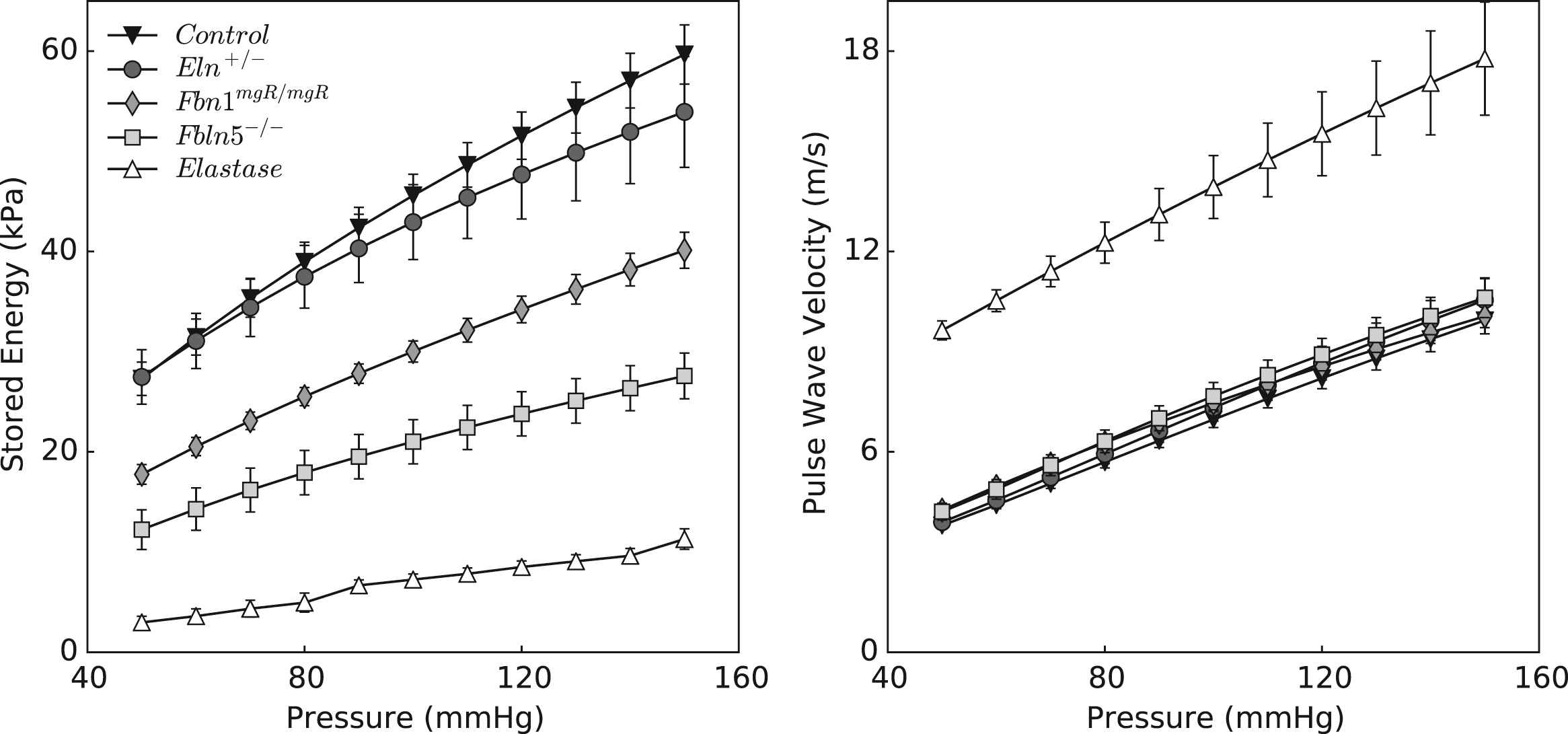
Stored energy (left panel) and computed local pulse wave velocity (right panel) plotted over a wide range of pressures. Trends shown in Table 1 and Fig. 5 for the energy storage at systolic pressures are valid at all pressures, confirming the sensitivity of this metric to distinguish different degrees of elastic fiber functionality. In comparison, pulse wave velocity calculated according to the Moens–Korteweg equation does not show the same descriptive trend.
Discussion
The human common carotid artery becomes progressively less distensible with increasing age,37 which appears to increase the risk of neurovascular-related conditions, including stroke.38 Distensibility and similarly pulse wave velocity, which reflect underlying structural stiffness, are convenient to measure and correlate with current or future clinical presentation, but they do not capture well the fundamental function of a central artery - storage and use of elastic energy. To glean increased insight, we measured pressure–diameter and axial force–length behaviors of murine common carotid arteries in vitro, which yielded information sufficient for quantifying stored energy as well as for computing common metrics of structural behavior (pulse wave velocity) and intrinsic material behavior (biaxial stress–strain relations and material stiffness). Despite being beyond the scope of the current study, combining information on biaxial mechanical properties gleaned in vitro with non-invasive assessments of distensibility in vivo should allow one to estimate the energy stored by the arterial wall under physiologic conditions while accounting for contributions of other factors that remain hard to quantify (e.g., effects of surrounding perivascular tissue on pressure-induced distention). We suggest, therefore, that a mechanistic understanding of the biomechanical factors underlying common prognostic indexes of cardiovascular function can only be achieved by accounting for the evolving multiaxial material properties of the arterial wall, whether during normal aging, hypertension, or progressive disease conditions.
Given the fundamental importance of elastic fibers to central artery structure and function, it is not surprising that many investigators have used mouse models (cf.33,39) to assess contributions of these fibers to arterial mechanics (e.g.,20,29,30,40–49). In particular, such studies generally report an increased stiffness due to loss of elastic fiber integrity, often inferred from pressure–diameter responses. The stiffness so inferred is necessarily a structural stiffness, which includes effects of both material stiffness and geometry. Our results are consistent with those of many prior studies, but emphasize further the importance of assessing the storage of elastic energy and biaxial material (intrinsic) stiffness. These quantities can best be evaluated from biaxial tests wherein both pressure and axial force are varied.23 Of the aforementioned studies, only four report complete biaxial data20,29,30,49 and ours is the first to employ a consistent approach to contrast biaxial behaviors across multiple mouse models to assess graded effects of lost elastic fiber integrity.
The present findings confirm recent results that mean circumferential material stiffness differs little in CCAs from controls and multiple mutant models, including muscular dystrophy, smooth muscle α-actin deficient, and aortic banding models.27 In contrast, other reports had suggested an increased circumferential modulus in some of these same mutants, which seemed intuitive. For example, there was an early report of a 4-fold increase in Young’s modulus in the descending thoracic aorta of the Fbn1mgR/mgR mouse40; this estimate was based on pulse wave velocity measured in vivo and inference of a modulus via the Moens–Korteweg equation. We adopted the opposite approach (see Appendix): we computed material stiffness directly from biaxial data collected in vitro and used circumferential values in the Moens–Korteweg relation to calculate pulse wave velocity (PWV). Similar to the circumferential stiffness, calculated values of PWV did not differ significantly between the genetically modified and control vessels considered here (Fig. 6). Although these calculations allowed a direct comparison of our results with prior in vivo findings, we must remember that the Moens–Korteweg equation yields a gross approximation because of its many simplifying assumptions, not the least of which is neglect of axial prestress. Given that axial mechanics can influence strongly the propagation of a pressure wave,50,51 recall that we found that a decreased axial stretch and stress (Table 1) helps to restore circumferential material stiffness toward normal levels (Fig. 3). It is thus important to account for the overall biaxial state. As noted previously, decreases in axial stretch appear to be early compensatory responses that allow an artery to unload itself both axially and circumferentially in response to diverse genetic or hemodynamic challenges.26 Although protective and often effective, extreme axial unloading can also render the vessel subject to lateral bending, which can manifest in vivo as tortuosity. Interestingly, varying degrees of tortuosity arise in central arteries of Eln+/−,42,45 Fbn1mgR/mgR,52 and Fbln5−/− mice.36 Consistent with these findings, significant lengthening occurs with acute elastase treatment,20 which decreases axial stretch dramatically (Table 1). There is a need to study further this important characteristic of compromised elastic fiber integrity.
Among the three mutant models studied herein, deletion of the elastin-associated glycoprotein fibulin-5 resulted in the most severe elastopathy. Fibulin-5 is thought to be involved in elastogenesis.18 Consequently, associated differences in mechanical behavior manifest early in the central arteries of these mice and, interestingly, progress little thereafter.49,53 In contrast, fibrillin-1 is thought to contribute to the long-term stability of elastic fibers, literally helping to protect these fibers against the mechanical fatigue that results from cyclic loading.17 That the biomechanical phenotype of Fbn1mgR/mgR CCAs, at 8 weeks of age, was mild may suggest that the appearance of breaks in elastic fibers in single cross-sections (Fig. 4) may not reflect a largely preserved underlying 3-D connectivity of these fibers. There is a need to consider two other key parameters, however: regional location and age. A more severe phenotype would be expected in the ascending aorta, which is subjected to full biaxial loading upon each beat of the heart, and with increasing age due to the accumulative effects of cyclic loading and earlier wave reflections resulting in increased central pulse pressures (cPP).54 Indeed, albeit at a lower rate, mechanical properties deteriorate with age in the aorta of another mouse model of Marfan syndrome (Fbn1C1039G/+, see43), which has an overall less severe aortic phenotype than the Fbn1mgR/mgR mouse.
Mechanical properties of central arteries have been studied extensively in the elastin haploinsufficient (Eln+/−) mouse.41,42,44,45,47 Consistent with prior reports, we found that, with respect to controls, Eln+/− carotids have a decreased arterial size (Fig. 1), increased number of elastic laminae (Fig. 4), and yet similar circumferential properties upon pressurization at their in vivo length (Table 1 and Fig. 2). In contrast with prior reports,42,45,47,55 however, no significant differences were observed in the in vivo axial stretch (Table 1 and Fig. 5), which was only slightly reduced with respect to controls. This discrepancy could be due to consistent methodological differences. For example, the in vivo axial stretch was previously inferred locally from motions of small affixed charcoal particles upon surgical removal of the vessel,42 noting that surgical access requires a repositioning of the neck from its native orientation. In contrast, we estimate this stretch (
Two of the major consequences of large artery stiffening in humans is an increased cPP, which increases the workload on the heart, and an increased propagation of undamped pressure waves to the microcirculation, which is thought to contribute to damage in low resistance end-organs.8,56,57 Increases in cPP have been observed in genetic models of altered elastic fiber integrity, particularly Eln+/− and Fbln5−/− mice (cf. Table 1), yet basic mechanisms underlying a high cPP could differ between these two models. There is mounting evidence, for example, that high cPP in elastin haploinsufficiency is due to increased systolic pressure.41,42,45,47,55,58 Although most of these investigations focused on central arteries, a recent study suggested that high blood pressure in Eln+/− mice is caused, in part, by structural and functional alterations of the resistance vasculature,59 including impaired endothelial function and increasing sensitivity to vasoconstriction in response to angiotensin II. The current findings of nearly normal axial stretches and resilience in Eln+/− CCAs, together with maintenance of circumferential material stiffness, supports the concept that high cPP may result primarily from increased peripheral resistances – and associated early wave reflections – in elastin insufficiency. The scenario seems to differ in mice lacking fibulin-5, in which loss of central artery function is also diffuse but more severe.24 Although high cPP was initially thought to be caused by increased systolic pressure,35 new evidence suggests that systolic pressure is maintained near normal levels while diastolic pressure is decreased (53 as well as unpublished observations from our laboratory on male mice). In terms of compensatory adaptations, a lower diastolic pressure shifts the response to a more compliant portion of the P–d curve, thus minimizing the impact of severe structural alterations on the distensibility of central arteries. By decreasing coronary perfusion pressure, however, a lower central diastolic pressure could yet have negative effects on diastolic function of the heart.53 These observations highlight the need to distinguish between central and peripheral contributions to high central pulse pressures.
Toward this end, we emphasize that computation of elastic energy storage is very appropriate for evaluating mechanical functionality of large (elastic) arteries because their primary function is to store and then use elastic energy to facilitate flow. In contrast, the primary functions of muscular arteries and arterioles are to regulate blood pressure and flow via vasoconstriction and vasodilatation, that is, changing smooth muscle contractility. Hence, different metrics focusing on active smooth muscle properties would be more appropriate for evaluating their mechanical competence. We did not contrast possible changes in active biaxial properties in the carotids because levels of in vivo basal tone are not known and could differ. Clearly, there is a pressing need for more attention to active properties.
In conclusion, our findings suggest that intramural cells may attempt to maintain values of circumferential stress and stiffness (Table 1) near those that are established via normal development while offsetting altered hemodynamic loads by changing structural stiffness. Maintenance of mean circumferential stress is consistent with a seminal observation that the tension per lamellar unit tends to be similar across mammalian species,60 despite very different arterial dimensions from mice to humans, as well as the provocative observation that circumferential material stiffness tends to be similar across invertebrates and vertebrates.61 Such maintenance is also consistent with a preferred micro-mechanical environment for cell sensing and regulation of matrix. There is, therefore, a need for more study of the implications of controlling matrix stiffness on cellular mechanobiological responses.62 Notwithstanding the important implications of maintaining circumferential stiffness and stress, there is yet a need to consider hemodynamic implications of increasing the structural stiffness and structural implications of axial unloading, which appeared to dictate the graded reduction in energy storage with loss of elastic fiber integrity and was the most striking finding herein (Figs. 4 and 5). Given that energy storage and usage is the most fundamental mechanical function of a large artery, and that damage to or degradation of elastic fibers is one of the hallmarks of arterial aging and hypertension, it is this irreversible biomechanical change that likely most affects cardiac function and flow augmentation. There is a need, therefore, to similarly investigate these characteristics along the aorta, which is responsible for the majority of systemic compliance.
Conflicts of interest statement
The authors declare no conflicts.
Acknowledgment
This work was funded, in part, by a grant from the
Appendix
Data from computer-controlled biaxial mechanical tests were used to calculate mean values of wall stress (σ) and stretch (λ) in both circumferential (ϑ) and axial (z) directions using standard formulae,63 namely
Whereas pressure–diameter (P–d) and axial force–length (f–l) data reveal the structural behavior of an artery (and depend on geometry), stress-stretch data (from which energy is calculated) reveal the intrinsic material behavior. Constitutive modeling of the arterial behavior was based on the following stored energy function23
Due to the nonlinear behavior of the carotid artery wall (Figs. 1 and 2), stiffness depends strongly on the combination of distending pressures and axial stretches imposed experimentally. Values of material stiffness useful in hemodynamic computations can thus be inferred by linearizing the stress-stretch behavior represented by the stored energy function W for any state of mechanical loading desired.64 Linearized elastic moduli were calculated as
Studies of the sensitivity of values of stored energy, material stiffness, and pulse wave velocity (PWV) to the value of pressure used in the calculation are shown in Figs. 3 and 6 by repeating the calculations for different values of distending pressure from 50 to 150 mmHg. All calculations in this work were based on the assumption that the axial stretch remains constant during the cardiac cycle and equal to the in vivo axial stretch
References
Cite this article
TY - JOUR AU - J. Ferruzzi AU - M.R. Bersi AU - R.P. Mecham AU - F. Ramirez AU - H. Yanagisawa AU - G. Tellides AU - J.D. Humphrey PY - 2016 DA - 2016/04/22 TI - Loss of elastic fiber integrity compromises common carotid artery function: Implications for vascular aging JO - Artery Research SP - 41 EP - 52 VL - 14 IS - C SN - 1876-4401 UR - https://doi.org/10.1016/j.artres.2016.04.001 DO - 10.1016/j.artres.2016.04.001 ID - Ferruzzi2016 ER -