
In silico-based Discovery of New Potential Drugs Targeting Severe Acute Respiratory Syndrome Coronavirus 2 Spike Glycoprotein
- DOI
- 10.2991/dsahmj.k.211103.001How to use a DOI?
- Keywords
- COVID-19; SARS-CoV-2; antiviral compounds; molecular docking; molecular dynamics simulations; spike glycoprotein
- Abstract
The SARS-CoV-2-induced novel coronavirus disease has become a global pandemic. As the latest coronavirus variants are even more infectious and deadly, its treatment is very difficult. Currently used drugs such as remdesivir, saquinavir, and chloroquine have several drawbacks. Recent studies have reported key proteins that could serve as drug targets. Amongst them, the spike (S) glycoprotein is an attractive drug target that plays a prominent role in viral binding and entry. With the aim of targeting and blocking the S protein, we designed a computational study for screening novel antiviral compounds. Molecular docking was used as a screening tool, and Molecular Dynamics (MD) simulations were used to further confirm the stability of ligand-bound complexes. The Asinex antiviral database was screened using a recently resolved S protein (PDB ID: 7C2L). The Schrodinger software suite was used for preparing the protein and ligand structures prior to performing the docking experiment. Based on the docking scores, antiviral compounds were screened and the docked complexes of top-performing hits were tested for complex stability and presence of molecular interactions using MD simulations. Finally, based on the nature of molecular interactions, six prospective hits— LAS 51389346, BDH 33920970, LAS 51389268, LAS 51389282, LAS 51389262, and LAS 51389430—were screened. Among these compounds, LAS 51389268, BDH 33920970, and LAS 51389268 showed consistent and strong binding with S glycoprotein, predicting them as prospective candidates for COVID-19 treatment. In conclusion, this study screened out novel prospective antiviral compounds that can intervene with virion entrance thereby being of potential use to treat COVID-19.
- Copyright
- © 2021 Dr. Sulaiman Al Habib Medical Group. Publishing services by Atlantis Press International B.V.
- Open Access
- This is an open access article distributed under the CC BY-NC 4.0 license (http://creativecommons.org/licenses/by-nc/4.0/).
1. INTRODUCTION
In December 2019, the new strain of coronavirus 2, known as COVID-19, alarmed the whole world. It started in the Chinese city of Wuhan and spread across ~187 countries because of its extremely contagious nature [1,2]. As of June 23, 2021, there have been an estimated 62,133,824 active cases, with approximately 3,894,282 deaths reported (https://www.coronalive.info/map). SARS-CoV-2 spreads mainly through respiratory secretions or droplet nuclei rather than through direct interaction [3]. The SARS-CoV-2 virus is classified in the order Coronavirales, which contains the family of coronaviruses [4]. It is a single-stranded, enveloped positive-sense RNA virus with a genome containing 29,891 nucleotides and encoding the 12 possible open reading frames responsible for the development of the viral structural and nonstructural proteins [5,6]. The spike glycoprotein of SARS-CoV-2 is critical for virus binding and entry [7]. It is a class I fusion protein located on the surface of the virus that helps the virus for its initial interaction with human Angiotensin-Converting Enzyme 2 (ACE2) receptors and then for fusing with the host cells [8]. Attachment is aided by the S1 subunit protein, which deploys its receptor-binding domain. As a result of interaction with the receptor, the protein undergoes a transition, from pre-fusion to post-fusion [9]. Association of ACE2 with the viral spike protein leads to viral infection. Inhibition of association between ACE2 and the viral spike protein with the help of an inhibitory ligand at the interface between ACE2 and the spike protein reduces the likelihood of infection [10,11]. Enzyme inhibitors that bind to either the ACE2 protein interface or the viral spike protein do not necessarily inhibit ACE2 enzyme activity.
Vaccine approaches have been developed through rigorous studies since the discovery of SARS-CoV-2 and the sequencing of its genome. Ten of these are in the final stages of clinical trials, three of which have been completed with positive results [12]. However, so far, preventive measures are the only mandatory means to limit the spread of infection among humans [13]. In Saudi Arabia, two vaccines are currently licensed and recommended for the prevention of COVID-19: Oxford–AstraZeneca COVID-19 vaccine and Pfizer–BioNTech vaccine (https://www.arabnews.com/node/1794826/saudi-arabia). Viral vector Oxford–AstraZeneca vaccine showed an efficacy of 76% after a first dose and maintained protection with the second dose [14]. Increase in efficacy (viz. 82%) was observed when the interdose interval was kept at 12 weeks or more. In the USA, Pfizer–BioNTech COVID-19 vaccine and Moderna COVID-19 vaccine (https://www.nytimes.com/2020/07/27/health/moderna-vaccine-covid.html) are currently used. The mRNA Pfizer–BioNTech vaccine offers an efficacy of 94.5% [15]. Moderna, another mRNA vaccine, showed a 94.5% efficacy [16]. Based on the shipping and storage conditions, Oxford–AstraZeneca vaccine holds an advantage, as it requires only normal refrigeration (4°C) whereas Pfizer vaccine requires ultralow temperature (−70°C). However, nearly 3 billion people in Africa, Asia, and Latin America will not be able to afford the COVID-19 vaccine until it is available years later [17]. Molecular docking has been used to advance the discovery of COVID-19 therapies [18]. However, currently approved therapies such as remdesivir, chloroquine, and dexamethasone, which inhibit the viral RNA-dependent RNA Polymerase (RdRp) enzyme, have many drawbacks, including a high incidence of side effects and drug–disease interactions [19–21]. The approach of computer-aided drug designing is a reliable and rational approach to expedite the process of drug development. The process of computational ligand screening not only reduces the need for costly laboratory work but also saves time, money, and resources, and removes the chances of end-stage drug failures. Accordingly, we have used computational approaches such as molecular docking-based compound screening and Molecular Dynamics (MD)-assisted binding check analysis for identifying novel COVID-19 agents. One of the recent computational approaches [10] investigated the potency of different ACE2 ligands to interfere with the recognition of the SARS-CoV-2 spike protein.
In this study, we identified novel hits of SARS-CoV-2 virus that can potentially act as an entry inhibitor for limiting its spread via S protein-based attachment. LAS 51389268, BDH 33920970, and LAS 51389268 have shown promising binding and stability through MD, indicating their possible usefulness in inhibiting COVID-19 entry. Based on our results we propose to carry further in vitro experimental studies for testing the therapeutic utility of LAS 51389268, BDH 33920970, and LAS 51389268 compounds in COVID treatment.
2. MATERIALS AND METHODS
2.1. Platform for Molecular Docking
The docking studies were carried out using the Schrodinger software (Maestro version 12.4, Schrodinger 2020-2; Schrodinger, LLC, New York, NY, USA) on a laptop workstation with Intel CORE i7, NVIDIA, GEFORCE, GTX.
2.2. Ligand Preparation
A total of 6827 purchasable antiviral compounds were retrieved from Asinex database (http://www.asinex.com/). The LigPrep module (Schrodinger) of Maestro version 12.4 [22] suite was used for ligand preparation, performing their geometry optimization for obtaining their minimum energy three-dimensional (3D) structures [23]. The Maestro builder and LigPrep were used to prepare the ligands and generate their 3D structures by adding hydrogen atoms, removing salt, and providing ionization at pH (7.4 ± 0.0) [24]. The OPLS3e force field and molecular mechanics-based standard energy function with Root-Mean-Square Deviation (RMSD) cut of 0.01 Å was used to select the low energy conformations [25].
2.3. Protein Preparation and Grid Generation
The crystal structure of the spike protein (7C2L.pdb) at 3.10 Å resolution was retrieved from the Protein Data Bank (http://www.rscb.org) [8,26,27]. The chain “A” of the spike protein was prepared using Protein Preparation Wizard tool of Schrödinger suite [28,29]. Correct bond orders and hydrogens were assigned to the protein [9]. The protein was prepared at physiological pH using PROPKA tool [30], and restrained minimization was performed using the OPLS3e force field [31].
Prior to selecting the top-ranked compounds, the characterization of drug-likeness and Absorption, Distribution, Metabolism, Excretion, Toxicity (ADMET) properties was employed using DruLiTo (http://www.niper.gov.in/pi_dev_tools/DruLiToWeb/DruLiTo_index.html) and PreADMET (http://www.bmdrc.org/preadmet/) server tools.
2.4. Molecular Docking
The default options of the receptor grid generation were used to identify the protein binding site. The centroid of the bound ligand was exploited for grid generation. Glide’s receptor grid box generation module was used to draw a cubic box of dimension 10 × 10 × 10 Å around the active site of the protein [32]. The better glide score and the presence of interactions with the binding site residues were considered as a criterion for ligand screening. First, the 6827 antiviral compounds were docked with the S protein in high-throughput virtual screening mode and based on the docking scores, the top 100 molecules were shortlisted. These compounds were then further screened using Standard Precision (SP) docking. Subsequently, Glide Extra Precision (XP) docking was performed to accurately predict the binding modes of potential hits with the spike protein of COVID-19 [33]. The biological evaluation of obtained hits needs to be done to further confirm their activity as potential COVID-19 inhibitors.
2.5. MD Simulations
The top-performing molecules from docking-based virtual screening were selected based on their XP docking scores [34] and the presence of critical molecular interactions. The ligand-bound complexes of these hits were submitted for MD simulations for estimating their binding stability. The Desmond MD tool was used and systems were prepared containing the protein–ligand complexes as immersed within the solvent box. The transferable intermolecular potential 3P water model was used as a system solvent. The orthorhombic simulation box (periodic boundary condition) was used keeping the buffer distance of at least 10 Å from the outer surface of the protein. The system was then neutralized at 150 mM salt concentration by adding Na+Cl− counterions maintaining the isosmotic conditions. Predetermined (default) equilibration protocol was used to prepare the systems prior to the production run. All MD simulations were performed using a thermostat of 300 K temperature and a barostat of 1.013 bar pressure. In total, 100-ns simulation was performed for each of the systems. Finally, the obtained simulation trajectories were analyzed using a simulation-interaction diagram and other analysis tools. All images were prepared using Maestro 12.4 (Schrödinger Release 2021-3: Maestro, Schrödinger, LLC, New York, NY, 2021), Discovery Studio Visualizer version 2019 [35], and gnuplot version 5.4 on Windows OS (http://www.gnuplot.info/).
3. RESULTS
3.1. Docking and Computational Simulations
The 6827 antiviral molecules from the Asinex antiviral library were screened using glide docking protocol against the spike glycoprotein target. Based on their binding at the estimated site and docking scores, compounds were ranked and screened. We meticulously analyzed the interactions of protein–ligand complexes from docking and further from MD simulation trajectories for stability checking. Combinedly results from both methods confirmed the presence of substantial and consistent ligand–binding interactions informing the stable complexes for top-ranked compounds. The XP docking results of the top 100 antiviral compounds demonstrated the ability to bind to the spike protein. A common trend of binding interactions and relatively comparable binding affinity data was witnessed for the top 13 compounds. They showed high binding affinity based on docking scores and Binding Energies (BEs), which range from −5.6 to −8.6 kcal/mol, compared with currently available antiviral drugs: remdesivir, saquinavir, lopinavir, paritaprevir, and chloroquine (Table 1). We also performed Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) calculations to accurately estimate the binding affinity (kcal/mol) of compounds. The consistent and quite comparable binding affinity was evident for the top-performing hits. The results of MD simulations showed that LAS 51389346, BDH 33920970, LAS 51389268, LAS 51389282, LAS 51389262, and LAS 51389430 hold stable interactions with the amino acid residues of the spike protein (mainly with the S2 domain) (Figure 1A–1F). The antiviral molecule LAS 51389346 holds hydrogen bond interactions with Lys 733, Phe 823, Leu 861, Pro 1057, and π–π stacking interactions with Hie, Pro 1057, 1058, Pro 863 residues. BDH 33920970 made H-bond interactions with Lys 733, Gln 774, Phe 823, and Asp 867, residues of the spike protein (S2 domain). LAS 51389268 showed Lys 733, Phe 823, Val 826, Leu 861, and Pro 1057 H-bonding. LAS 51389282 exhibited H-bonding interactions with almost similar residues of the spike protein (S2) and π–π stacking interaction with Hie 1058 residue. LAS 51389262 made Asp 867, Leu 861, Val 860, Lys 733, Met 731, Pro 1057, and Phe 823 residues. LAS 51389430 made Asp 867, Leu 861, Val 860, Lys 733, and Phe 823 residues, and π–π stacking interaction with Pro 1057, His 1058 residues. Overall, these six top-ranked compounds hold quite comparable and binding interactions with the spike protein (S2) as shown in 2D and 3D binding poses (Figure 1A–1F). Moreover, based on molecular docking, MD simulations, and MM/GBSA calculations, the top six compounds showed binding at protein residues such as Lys 733, Phe 823, Leu 861, Asp 867, Pro 1057, and Hie 1058, forming a well-defined binding pocket at the S2 domain of the spike protein, as shown in Figure 2.
| Sr. no. | ID | Docking score | Binding affinity (kcal/mol) | Amino acid residues interacting with Asinex antiviral compounds |
|---|---|---|---|---|
| 1 | LAS 51389262 | −7.9 | −41.93 | Lys 733, Leu 861, Pro 1057, Asp 867, Phe 823, Hie 1058 |
| 2 | LAS 51389268 | −7.8 | −40.08 | Lys 733, Leu 861, Thr 732, Val 826, Phe 823, Asp 867, Pro 1057, Hie 1058 |
| 3 | LAS 51389274 | −8.6 | −41.03 | Lys 733, Phe 823, Val 826, Leu 861, Asp 867, Pro 1057, Hie 1058 |
| 4 | LAS 51389430 | −7.5 | −37.56 | Lys 733, Leu 861, Asp 867, Phe 823 |
| 5 | LAS 51389270 | −7.4 | −41.28 | Lys 733, Phe 823, Leu 861, Val 826, Asp 867, Hie 1058 |
| 6 | LAS 51389346 | −7.3 | −42.42 | Lys 733, Phe 823, Leu 861, Pro 1057, Hie 1058 |
| 7 | LAS 51389282 | −6.7 | −38.96 | Phe 823, Asp 867, Leu 861, Lys 733 |
| 8 | LAS 51389382 | −6.5 | −38.46 | Lys 733, Leu 861, Phe 823 |
| 9 | LAS 51389302 | −6.3 | −43.25 | Lys 733, Leu 861, Phe 823 |
| 10 | LAS 51153877 | −6.1 | −23.07 | Thr 778, Ser 730, Asp 867 |
| 11 | BDH 33920970 | −6.2 | −21.85 | Phe 823, Asp 867, Asp 775 and Gln 774 |
| 12 | BDI 34016890 | −6.0 | −35.24 | Lys 733, Leu 861, Asp 867, Thr 778, Pro 1057 |
| 13 | LAS 51389452 | −5.6 | −43.29 | Thr 827, Lys 733, Asp 867, Leu 861 |
| 14 | CID:121304016 | −5.2 | −43.95 | Asp 867, Hie 1058 |
| Remdesivir | ||||
| 15 | CID: 441243 | −3.8 | −34.71 | Asp 867 |
| Saquinavir | ||||
| 16 | CID: 92727 | −3.2 | −50.33 | Asp 867 |
| Lopinavir | ||||
| 17 | CID: 45110509 | −2.9 | −41.06 | – |
| Paritaprevir | ||||
| 18 | CID: 2719 | −2.7 | −16.45 | – |
| Chloroquine |
Top antiviral compounds (http://www.asinex.com/) docked against COVID-19 spike protein

Docking pose for molecules: (A) LAS 51389346, (B) BDH 33920970, (C) LAS 51389268, (D) LAS 51389282, (E) LAS 51389262, and (F) LAS 51389430 with 7C2L. The H-bonds are depicted in green lines; carbon-mediated H-bonds in light green lines, whereas π–π interactions are depicted in purple lines, and π-cation interactions are depicted with light purple lines.

The novel binding site of spike glycoprotein at the S2 domain; both Pro1057 and His1058 help the proper placement of ring systems, in which the earlier forms the cavity for placing the hydroxyphenyl ring whereas the latter forms the π–π stacking interactions with ligand pentameric ring (middle). The methoxyphenol ring complimentarily fits inside nicely interacting residues Lys 733, Leu 861, and Val 860.
The binding site residues are mainly from the S2 subunit (686–1273 residues), whereas S1 subunit N-terminal residues (14–685) do not contribute to forming this cavity. The S2 region consists of the fusion peptide (FP) (788–806 residues), Heptapeptide Repeat sequence 1 (HR1) (912–984 residues), HR2 (1163–1213 residues), transmembrane (TM) domain (1213–1237 residues), and cytoplasm domain (1237–1273 residues) [36]. During viral entry, protein priming by cellular proteases cleaves the S1/S2 domains and formed S2 allows the membrane binding of viral protein. Our identified binding site lies in between HR1 and HR2 sequences and involves the residues nearby to TM domain. The S1 region facilitates ACE receptor binding whereas the S2 region is responsible for membrane binding. The binding of ligands at the S2 site is likely to inhibit the membrane binding of the spike protein thereby inhibiting the viral entry to the host cell.
3.2. Molecular Dynamic Simulation
Molecular dynamics simulations were performed with the objective of checking the stability of protein–ligand bound complexes. The six top compounds as screened from glide-docking were further considered for the dynamic stability check. The selected protein–ligand bound poses of LAS 51389346, BDH 33920970, LAS 51389268, LAS 51389282, LAS 51389262, and LAS 51389430 were used as a starting point of this study. Dynamics of protein and ligand structures in the bound complex were tracked using 100-ns MD simulations. The RMSD analysis of LAS 51389346 clearly exhibited the stable residence of ligand for the entire 100-ns simulation (Figure 3A and 3B). The protein RMSD is quite stable whereas the RMSD of ligand fitted over the protein shows an initial surge of up to 30 ns followed by stability for the remainder of the simulation. The RMSD of protein was very much stable around 20 Å. In Supplementary Figure S1B, during MD simulations, LAS 51389346 bound to the S2 domain displayed both polar (water bridges) and nonpolar (hydrophobic) interactions via the amino acid residues Lys 733, Phe 823, Asp 867, Leu 861, Pro 1057, and π-cation stacking interaction with Hie 1058. The amino acid residue Asp 867 made water bridged as well as direct hydrogen bond interaction with LAS 51389346 ligand. Residues Pro 1057 and Pro 863 showed 50% hydrophobic interactions, and Phe 823 showed 30% hydrophobic interactions (Supplementary Figure S1B) during the simulations. The interaction with residue Leu 861 was consistent throughout the simulation, indicating the presence of this strong interaction as shown in Figure 4. Similarly, Asp 867 also showed such consistent interaction. Moreover, Lys 733 showed very strong and consistent interaction and Hie 1058 showed consistent interaction with all ligands. The RMSD of the superimposed spike protein (72CL) aligned with the ligand LAS 51389346. Figure 2 shows a large drift at the beginning of the simulation at 20 ns, indicating high protein fluctuations.

The Root-Mean-Square Deviation (RMSD) plots. Protein RMSD (A) and ligand RMSD fitted against the protein structure viz., ligand with respect to protein (B) is shown for LAS 51389346, BDH 33920970, LAS 51389268, LAS 51389282, LAS 51389262, and LAS 51389430. The RMSD deviations are calculated against the starting structure of the complex over the period of 100-ns simulation shown as 1000 frames.
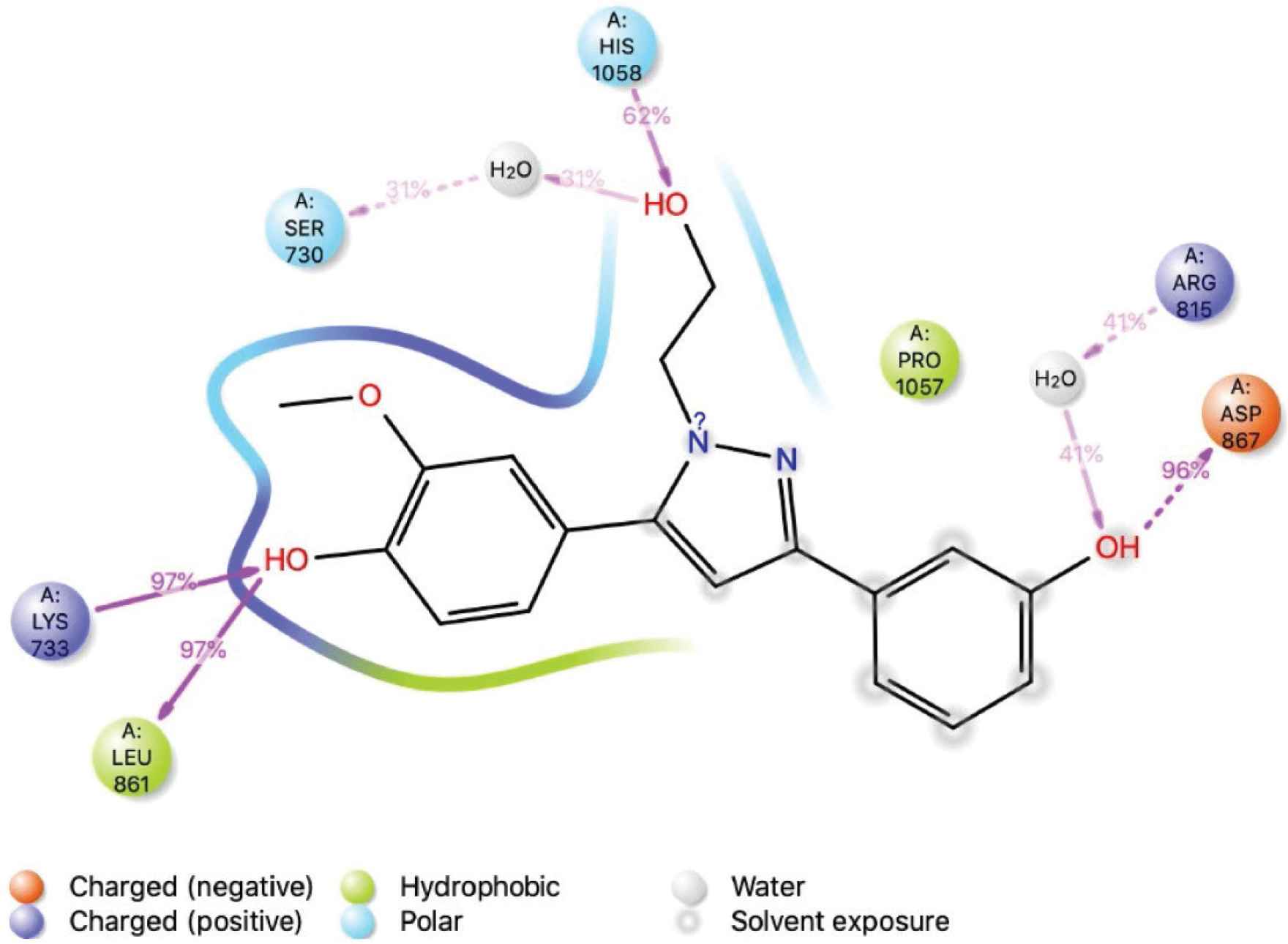
The interactions of amino acid residues of spike protein (S2) with ligand LAS 51389346. LAS 51389346 binding interaction with the residues of the protein during the simulation. More than 30% of the interactions shown occur during the simulation period.
We also analyzed the ligand positioning using RMSD, Molecular Surface Area (MOLSA), Solvent Accessible Surface Area (SASA), and Polar Surface Area (PSA) for confirming the nature of ligand binding for the individual ligands (Supplementary data). The RMSD of the ligand aligned against the starting snapshot of the ligand was very stable and showed no changes in position or size (radius), indicating that LAS 51389346 remained stably bound in the binding site. MOLSA, SASA, and PSA of the ligand were also very stable and showed no changes (Figure 5). Although the RMSD of the spike protein shown in Figure 2 was very high and deviating, the ligand properties were very consistent and showed more than 90% (very stable) interactions (Figure 3). As well as the ligand, LAS 51389346 molecule showed very promising stability and could be a very potent inhibitor for the spike protein of COVID-19 (Supplementary data).
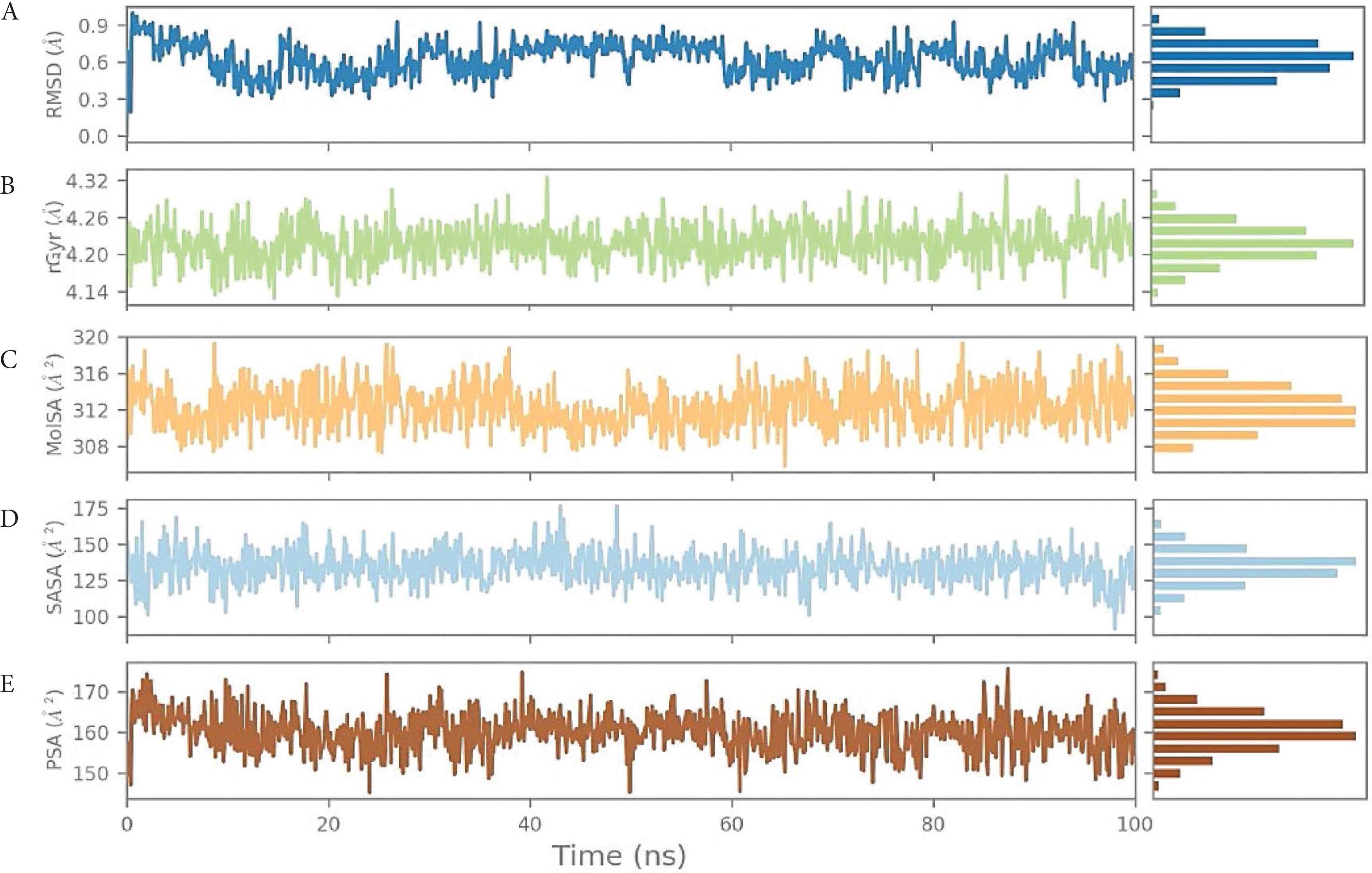
(A) Root-Mean-Square Deviation (RMSD) of the ligand LAS 51389346 with respect to the reference time (tref) (the first frame is usually used because the reference is considered to be the zero time) (Supplementary data). (B) rGyr: Measures the “expansion” of a ligand and is equivalent to its principal moment of inertia. (C) MOLSA: molecular surface area calculation with 1.4 Å probe radius. The surface area of this is equal to a van der Waals. (D) SASA: surface area of a molecule accessible to a water molecule. (E) PSA: surface area of a molecule accessible by the solvent, supported only by oxygen and nitrogen atoms.
The RMSD of the BDH 33920970–protein complex showed larger fluctuations compared with LAS 51389346, which showed an increasing trend in the first 20-ns run, and further continued increase until the 60-ns run, but afterward between 60 and 100 ns, the protein remained constant within the window of 4 Å change. The ligand showed significant structural shifts between 20 and 32 Å, relatively changing its orientation inside the binding site. Until the end of the simulation, the RMSD of the ligand–protein complex ranged near 30 Å, suggesting comparatively less stable binding of BDH 33920970 (BDH 33920970 is one of the antiviral compounds presnt in the asinex antiviral database [http://stock.asinex.com/compounds]). During dynamics, residues Asp 867, Leu 865, Asp 775, Thr 778, and Phe 823 of the spike protein (S2) formed hydrogen bonds (30–100%) with the ligand BDH 33920970 as shown in Supplementary Figure S2B. This molecule displayed hydrogen bonding, as well as water bridges and hydrophobic interactions. The amino acid residue Asp 867 also showed both water-bridged and direct hydrogen bonding with the ligand at more than 100% (Figure 6). RMSD, MOLSA, SASA, and PSA of the ligand was stable and did not show any changes in position or size (radius) (Supplementary data), indicating its stable binding at the S2 site (Figure 7).

The interactions of amino acid residues of spike protein (S2) with ligand BDH 33920970. BDH 33920970 binding interaction with the residues of the protein during simulation. More than 30% of the interactions shown occur during the simulation period.
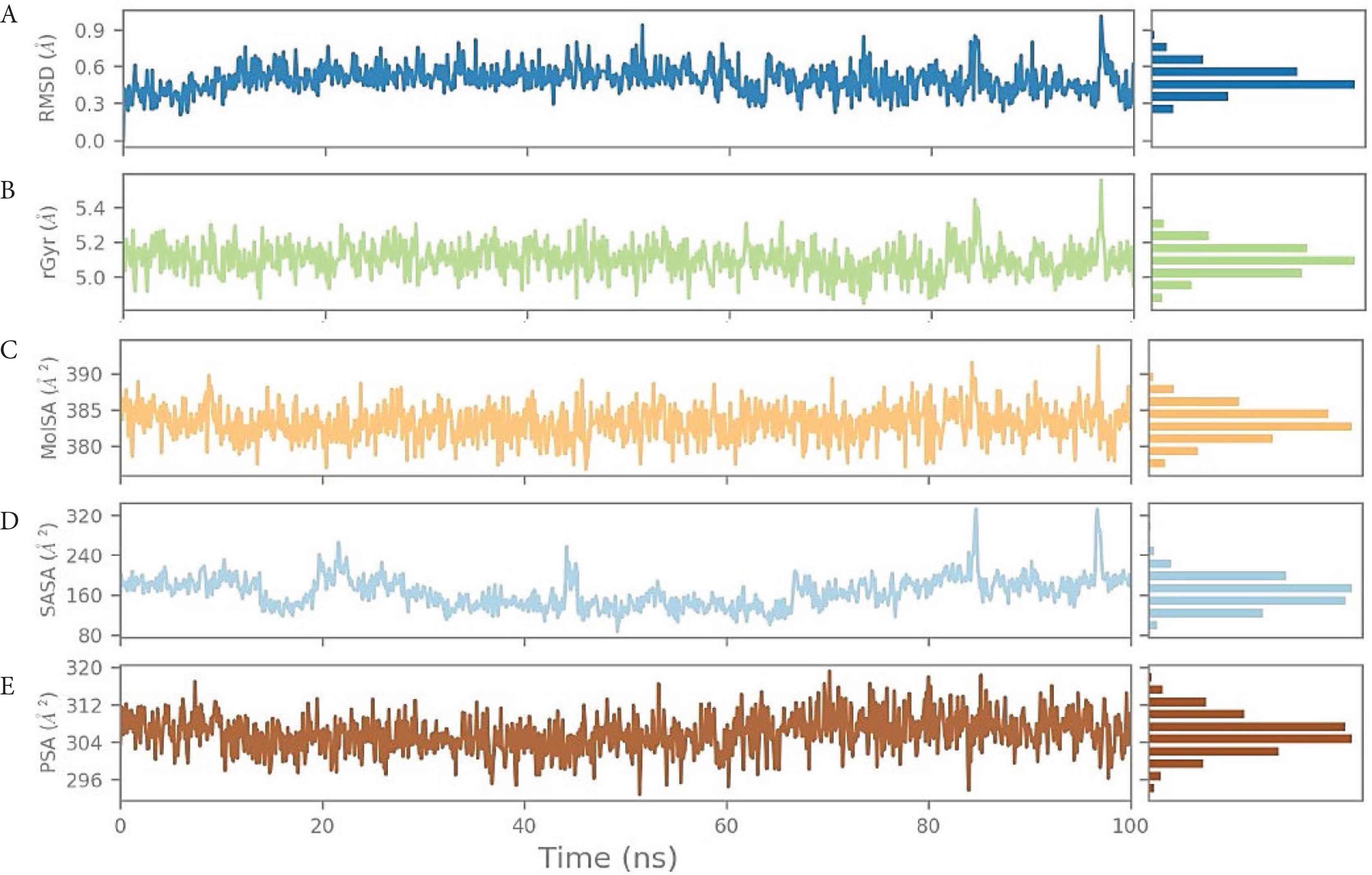
(A) Root-Mean-Square Deviation (RMSD) of the ligand BDH 33920970 with respect to the reference time (tref) (the first frame is usually used because the reference is considered to be the zero time) (Supplementary data). (B) rGyr: measures the “expansion” of a ligand and is equivalent to its principal moment of inertia. (C) MOLSA: molecular surface area calculation with 1.4 Å probe radius. The surface area of this is equal to a van der Waals. (D) SASA: surface area of a molecule accessible to a water molecule. (E) PSA: surface area of a molecule accessible by the solvent, supported only by oxygen and nitrogen atoms.
Consistent binding stability was observed for the protein and ligand complex of LAS 51389268 as shown in Figure 3. The protein RMSD of LAS 51389268 is quite comparable with BDH 33920970, showing initial fluctuations prior to the 40-ns run, and later protein starts to equilibrate. The RMSD for ligand–protein structure remained under 25 Å until the end of simulation.
In Figure 8, although the decline in the number of amino acid interactions, a combination of hydrogen bonds, and polar and nonpolar hydrophobic interactions was seen during simulations for Pro 1057, Leu 861, Lys 733, and Phe 823. In Supplementary Figure S3B, the panel shows that residues Lys 733, Leu 861, and Pro 1057 strongly interacted with ligand, as represented by a darker shade of orange, according to the scale (on the right side of the plot). Residues Leu 861, Lys 733, and Pro 1057 formed hydrogen bonding interactions with LAS 51389268 at approximately 98%, 93%, and 87%, respectively, indicating a strong binding interaction. Bridged hydrogen bonding was observed between the ligand and residues Asn 867, Leu 865, and Arg 815. A π–π interaction was observed for Hie 1058 (63%) (Supplementary Figure S3C). RMSD, MOLSA, SASA, and PSA of the LAS 51389268 molecule were very stable and showed no changes in position or size (radius), indicating stable binding of LAS 51389268 at the binding site (Figure 9).

The interactions of amino acid residues of spike protein (S2) with ligand LAS 51389268. The binding interaction with the residues of the protein during the simulation. More than 30% of the interactions shown occur during the simulation period.

(A) Root-Mean-Square Deviation (RMSD) of the ligand LAS 51389268 with respect to the reference time (tref) (the first frame is usually used because the reference is considered to be the zero time) (Supplementary data). (B) rGyr: measures the “expansion” of a ligand and is equivalent to its principal moment of inertia. (C) MOLSA: molecular surface area calculation with 1.4 Å probe radius. The surface area of this is equal to a van der Waals. (D) SASA: surface area of a molecule accessible to a water molecule. (E) PSA: surface area of a molecule accessible by the solvent, supported only by oxygen and nitrogen atoms.
In summary, although initially the RMSD (for both protein and ligand) ranged higher for complexes, later it became stable after ~30 ns and remained varying within the narrow window of ~2–4 Å, indicating the stable nature of complexes containing the bound ligand.
4. DISCUSSION
The spike protein is a homotrimer that covers the surface of coronavirus and plays a central role in viral entry, and consists of two subunits, S1 and S2 [37]. S1 binds to the cellular receptor via its Receptor Binding Domain (RBD), whereupon the S2 domain undergoes a series of conformational changes allowing the FP to integrate inside the host cell membrane [38–40].
A recent computational docking study revealed the role of human Toll-like receptors (TLRs)—that is, TLR4—against S protein, predicting it might affect the virus-induced inflammatory consequences associated with COVID-19 [41]. Therefore, the authors recommended the designing of competitive TLR4-antagonists for selective targeting of TLR4–spike protein interaction. They also observed the strong binding of SARS-CoV-2 S protein with ACE2 receptors from both human and bat origin (as phylogenetically close), indicating its usefulness in adopting S protein-, ACE2-, or TLR-guided intervention strategy against COVID-19 disease [42]. Binding of ivermectin was studied against the key proteins of SARS-CoV-2 pathogenesis using molecular docking and MD simulations [43]. This study predicted the blocking actions of ivermectin against viral replicase, protease, and human transmembrane serine protease 2 (TMPRSS2) supporting its utility in COVID treatment. With the help of molecular modeling and MD simulations, the functional significance of various TLRs (viz., TLR3, TLR7, TLR8, and TLR9) was estimated in terms of their comparative binding with SARS-CoV-2 mRNA [44]. This work clearly depicted the role of TLR3, TLR7, and TLR9 in COVID-19 immunobiology, and revealed the potential of these novel SARS-CoV-2 targets. Choudhury et al. [45] have nicely commented about the use of chemotherapy versus immunotherapy in combating the novel COVID-19 infection [45]. The COVID outbreak has significantly affected heart functioning; thus, the therapeutic choices for myocardial inflammation caused by COVID-19 infection also need to be considered seriously [46].
We utilized a similar computational protocol and planned for a systematic drug designing study to identify the novel lead compounds targeting the SARS-CoV-2 spike protein. We performed molecular docking-based screening of a total of 6827 antiviral compounds from the Asinex database (http://www.asinex.com). After docking the best interacting compounds were screened out for further studies. Computational modeling was performed using Maestro interface (Schrodinger Suite), which offers an efficient drug designing platform to obtain fruitful outcomes. From our data, we can state that the molecular docking-based screening approach can be effectively used to explore alternative uses of existing drugs for repurposing at other therapeutic targets.
In the present study, after the preliminary screening of 6827 antiviral compounds, we identified six antiviral compounds that have the potential to bind at the S2 site of the spike protein. Based on glide scores, MD simulations, MM/GBSA calculations, and visual inspection of data, the top-ranking six compounds are estimated to bind at the S2 region, and can thereby intervene in the viral entry to the host cell by inhibiting the membrane binding of S′. Their BEs range from −5.61 to −8.9 kcal/mol. The drug-likeness screening of the top six compounds follows Lipinski’s rule of five (RO5) and Veber’s rule. Moreover, ADMET predictions showed no Pgp inhibition and are relatively safer, encouraging their likelihood as future drugs. Similarly, other groups [47] have identified several compounds from fungi that exhibit optimal binding at spike proteins, where BE ranged from −9.1 to −10.5 kcal/mol as obtained using molecular docking. Several compounds fulfilled the requirements for binding with S protein, of which quinazoline B, rubrolide S, and 11α-dehydroxyisoterreulactone A were identified as the most potent inhibitors, with BEs of −10.5, −10.3, and −10.2 kcal/mol, respectively.
Based on our screening and MD simulation data for antiviral database compounds, clearly it is well within reach to inhibit the entry and subsequent viral infection of COVID-19 to the host cell by targeting the S2 pocket of the spike protein. Even though we did not track any significant conformational changes from the short 100-ns MD simulations, the stability, and the interaction profiles of database compounds were clearly established. The novel S2 binding site and the identified scaffold of antiviral compounds warrant further experimental validation.
Based on the results of our MD simulations and interactions analysis, we propose three new antiviral compounds—LAS 51389346 {4-[1-(2-hydroxyethyl)-3-(3-hydroxyphenyl)-1H-pyrazol-5-yl]-2-methoxyphenol}, BDH 33920970 {4-(3-aminopropoxy)-N-[(1R,2R,3R,4R)-4-(2,4-dioxo-1,2,3,4-tetrahydropyrimidin-1-yl)-2,3-dihydroxycyclohexyl]benzamide}, and LAS 51389268 {5-[5-(4-hydroxy-3-methoxyphenyl)-1-(2-hydroxyethyl)-1H-pyrazol-3-yl]benzene-1,2,4-triol}—as potential starting leads that can target the S2 region of the spike protein and thereby inhibit the entry of COVID-19 virus. The more robust and stable LAS 51389346-S2 complex as observed through MD simulations appears to be the most promising lead molecule to start with further experimental investigations related to its anti-COVID-19 activity. To our knowledge, the antiviral compounds identified in the current study were not presented in the literature prior to the time of publication, and we are the first to identify them as novel anti-COVID-19 compounds. We are currently investigating these compounds against other SARS-CoV-2 proteins such as Mpro and RdRp enzymes.
5. CONCLUSION
Because COVID-19 has high transmissibility, and as there are very limited existing drugs or vaccines to counteract it, we encourage the use of computational approaches for finding new therapeutic compounds such as those presented in this in silico study. Despite previous attempts, our research focused on the antiviral library of Asinex to find possible inhibitors of spike glycoprotein (S2) domain. There have been no previous records of using such inhibitors against the spike-binding S2 domain for interrupting virus–cell membrane fusion of COVID-19. Out of the 6827 virtually screened antiviral compounds, three antiviral compounds—LAS 51389346, BDH 33920970, and LAS 51389268—were identified as potent hits with possible inhibitory activity against the spike protein in SARS-CoV-2. The complexes formed between the top-ranked ligands and the spike protein (S2) in MD simulations were dynamically stable and possess significant binding as observed from binding free energy data. Although these molecules are candidates for further discovery of multitargeting against SARS-CoV-2, they can be used as models or prototypes. This work serves as a cornerstone for future COVID-19 disease studies and for the discovery and development of new multitarget compounds.
CONFLICTS OF INTEREST
The authors declare they have no conflicts of interest.
AUTHORS’ CONTRIBUTION
All authors contributed equally to drafting and revising the manuscript. They approved the final version for publication.
ACKNOWLEDGMENTS
The authors are grateful to Dr Sulaiman Al Habib Medical Services Group (Saudi Arabia) for providing the financial resources required for this study by financing an initiative to accelerate scientific and medical research on the emerging coronavirus (COVID-19). In addition, the authors acknowledge the efforts of the Department of Pharmaceutics, Manipal College of Pharmaceutical Sciences, which assisted with the computer simulations.
SUPPLEMENTARY MATERIALS
Supplementary information associated with this article can be found in the online version at https://drive.google.com/drive/folders/1ee3umnqFPXWnxySY49i5D84ZIsXQWsYl. Also available at
Footnotes
Data availability statement: The datasets presented in this study can be found online at https://drive.google.com/drive/folders/1ee3umnqFPXWnxySY49i5D84ZIsXQWsYl and https://doi.org/10.2991/dsahmj.k.211103.001
REFERENCES
Cite this article
TY - JOUR AU - Hayam Abdelkader AU - Mahmoud Rifaat AU - Mohammed Baghdadi AU - Nariman Sindi AU - Radwa Rifaat PY - 2021 DA - 2021/11/15 TI - In silico-based Discovery of New Potential Drugs Targeting Severe Acute Respiratory Syndrome Coronavirus 2 Spike Glycoprotein JO - Dr. Sulaiman Al Habib Medical Journal SP - 168 EP - 178 VL - 3 IS - 4 SN - 2590-3349 UR - https://doi.org/10.2991/dsahmj.k.211103.001 DO - 10.2991/dsahmj.k.211103.001 ID - Abdelkader2021 ER -