
The Many Faces of Cytokine Release Syndrome-Related Coagulopathy
 , John Doran
, John Doran- DOI
- 10.2991/chi.k.210117.001How to use a DOI?
- Keywords
- Cytokine; Coagulopathy; CAR T cell; Hemophagocytic lymphohistocytosis; COVID-19
- Abstract
Cytokine release syndrome (CRS) has been increasingly recognized in various conditions including the coronavirus disease 2019 (COVID-19). It is not only associated with systemic inflammatory symptoms, but also hematological complications such as coagulopathy. CRS can affect various components of the coagulation pathway, including the endothelial cells, platelets, coagulation cascade, and fibrinolytic system. Different causes of CRS, such as primary hemophagocytic lymphohistocytosis (HLH), chimeric antigen receptor (CAR) T-cell therapy, and COVID-19, have different cytokine profiles and coagulopathy presentations, with microvascular thrombosis surfacing as a common pathology. HLH shares many features with severe CRS, and is characterized by severe consumptive coagulopathy, frequent disseminated intravascular coagulation and an increased bleeding risk. CAR T-cell therapy is characterized by frequent and mild consumptive coagulopathy, as well as an increased risk of thrombosis. While consumptive coagulopathy is rare in COVID-19, it is associated with an increased thrombotic risk. The differences can be explained by the severity of CRS and underlying conditions associated with coagulopathy. Various treatments, including cytokine inhibitors, plasma exchange, Janus kinases inhibitors, complement blockade, and corticosteroids are being studied to mitigate CRS-related coagulopathy.
- Copyright
- © 2021 International Academy for Clinical Hematology. Publishing services by Atlantis Press International B.V.
- Open Access
- This is an open access article distributed under the CC BY-NC 4.0 license (http://creativecommons.org/licenses/by-nc/4.0/).
1. INTRODUCTION
Cytokine release syndrome (CRS), or cytokine storm, is an acute inflammatory syndrome caused by the activation of immune cells and release of proinflammatory cytokines. Starting as a fever, it can rapidly progress to hypotension, hypoxia, and end-organ dysfunction. Since the advent of chimeric antigen receptor (CAR) T-cell therapy, our understanding of the pathophysiology and treatment of CRS has greatly progressed. Initially described in patients receiving muromonab-CD3 [1], CRS is now commonly encountered in primary hemophagocytic lymphohistocytosis (HLH), bi-specific T-cell engager (BiTE) therapy, CAR T-cell therapy, haploidentical hematopoietic stem cell transplantation (haplo-HSCT) with post-transplantation cyclophosphamide (PTCy), as well as the coronavirus disease 19 (COVID-19) [2]. In addition to causing systemic inflammatory response, CRS is also linked to hematological toxicities such as coagulopathy.
The association between inflammation and thrombosis has been increasingly recognized in recent years. The term “thromboinflammation” was first used by Blair et al. [3] in 2009 to describe the activation of Toll-like receptors on platelets, leading to both inflammatory response and thrombosis. Since then, a growing body of literature supports the association between thrombosis and various inflammatory states — infection, autoimmune disorders, viperid snake envenomation, major trauma, and ischemia/reperfusion injury. This review will focus on the different presentations of coagulopathy associated with three different conditions leading to a CRS: primary HLH, CAR T-cell therapy, and COVID-19. These three entities are, so far, the best studied. Here, we summarize the pathogenesis, presentations, as well as management of CRS-related coagulopathy.
2. MECHANISTIC ASSOCIATION BETWEEN CRS AND COAGULATION
Cytokine release syndrome is characterized by the elevation of various cytokines, most noticeably interleukin-6 (IL-6), IL-1, interferon-gamma (IFN-γ), and tumor necrosis factor alpha (TNF-α). Other cytokines, including IL-8, IL-10, monocyte chemoattrac-tant protein-1 (MCP-1), and granulocyte-macrophage colony-stimulating factor (GM-CSF) are also consistently elevated [4]. The formation of thrombosis occurs in two steps — primary hemostasis which involves the crosstalk between the endothelial cells and platelets, and secondary hemostasis which involves the coagulation cascade. Different cytokines can affect different components of the thrombosis formation process, leading to a hypercoagulable state (Figure 1).
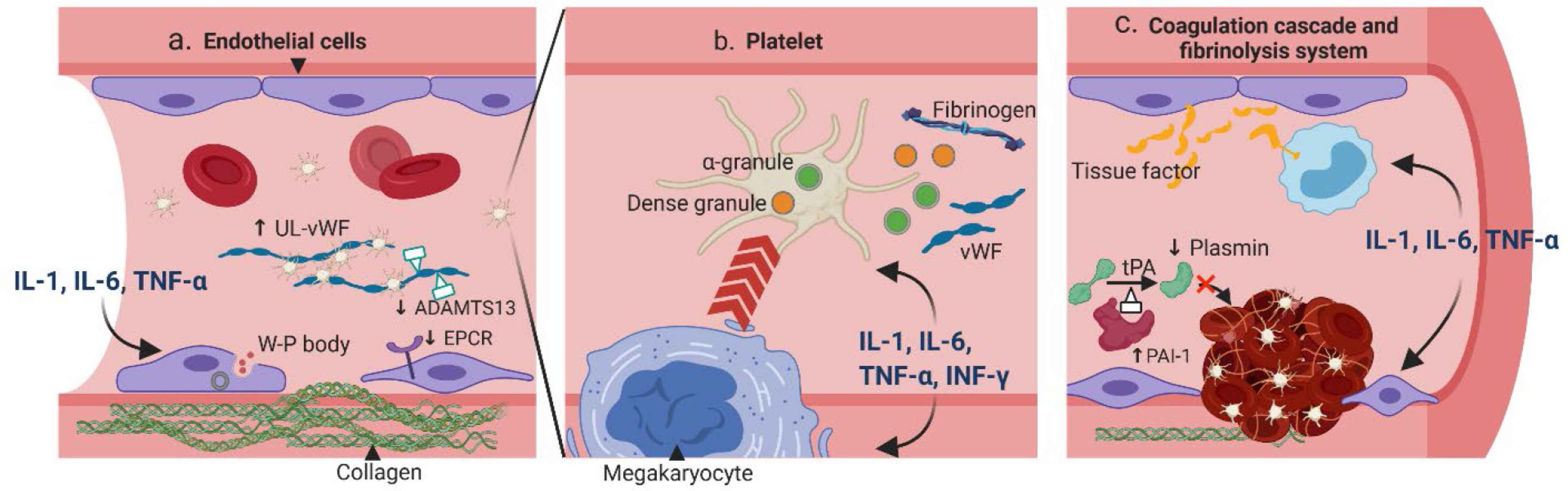
Pathogenesis of CRS-related coagulopathy. (a) Pro-inflammatory cytokines IL-1, IL-6 and TNF-α can stimulate endothelial cells (ECs) to secrete Weibel–Palade (W–P) body, which contains ultra-large von Willebrand factor (UL-vWF) and P-selection; both are essential for platelet attachment. Moreover, the cytoskeletons of ECs are rearranged to expose the procoagulant collagen. The expressions of anticoagulant proteins such as the endothelial protein C receptors (EPCR) and ADAMTS-13 are also downregulated. (b) Cytokines IL-1, IL-6, TNF-α and IFN-γ can affect the quantity and quality of platelets. The megakaryocyte maturation is accelerated resulting in more platelets. These cytokines can also stimulate the release of dense granules and α-granules, which contain essential coagulation substrates such as the fibrinogen and vWF. (c) Cytokines IL-1, IL-6, and TNF-α can increase the expression of tissue factor from ECs and on monocytes. In addition, the fibrinolytic system is inhibited with increased expression of plasminogen activator inhibitor type I (PAI-1), which can deactivate tPA and result in less plasmin. ADAMTS, a disintegrin and metalloproteinase with thrombospondin motifs; IFN, interferon; tPA, tissue plasminogen activator.
2.1. Effects of Cytokines on Endothelial Cells
The endothelium covers the lumen of the entire venous and arterial system. Resting endothelial cells (ECs) help maintain homeostasis by preventing platelet adhesion, decreasing inflammation, and releasing natural anticoagulants [5]. However, upon exposure to inflammatory stimuli, ECs can rapidly transform the surrounding environment into a procoagulant state, through exocytosis of procoagulant particles, breakdown of the endothelial barrier, and down-regulation of the anticoagulant pathways [6]. This process, also known as the type II endothelial activation or delayed sustained activation, is the initial step of inflammation-related coagulation [7]. More specifically, cytokines such as IL-1, IL-6 and TNF-α can activate the MAPK/NF-κB pathway within the ECs, leading to synthesis and release of procoagulant particles such as the Weibel–Palade bodies [8–10]. The particles contain ultra-large von Willebrand factor (vWF) and P-selectin, which can together form a network with other coagulant factors, platelets and inflammatory cells. In addition, the signaling pathway activation can also lead to reorganization of the ECs’ cytoskeleton [11] and loss of tight junctions [12], resulting in exposure of the highly coagulant collagen. Moreover, the natural anticoagulant pathways are inhibited when cytokines are present, through mechanisms such as decreased expression of endothelial protein C receptors [13] or inhibited synthesis of a disintegrin and metalloproteinase with thrombospondin motifs 13 (ADAMTS-13) [14].
2.2. Effects of Cytokines on Platelets
Platelets not only serve as building blocks of the hemostatic plug, but also act as gatekeepers of the vascular wall to help preserve vascular integrity during inflammation in a non-aggregated form. Cytokines can affect platelets both quantitatively and qualitatively. In vitro studies found that IL-6 can promote the maturation of megakaryocytes [15], and injection of IL-6 in humans was associated with an increased platelet count [16]. On the other hand, IL-1 administration can not only promote megakaryocyte differentiation, but also induce its rupture, leading to immediate release of platelets into the circulation [17]. Other cytokines, such as the TNF-α and IFN-γ, have also been shown to increase the platelet production [18]. Moreover, cytokines can regulate the secretion of platelet granules. The dense granules of platelets are filled with small molecules such as adenosine diphosphate (ADP), calcium, and polyphosphate (polyP), whereas the α-granules contain coagulation factors such as the vWF, fibrinogen (factor I), factor V, XI and XIII [19]. Both granules contribute to the platelet aggregation and the coagulation cascade. Upon inflammation, IFN-γ and IL-1 can enhance platelet dense granule secretion [20], while IL-2 can attenuate α-granules secretion [21]. In addition, activated platelets can reversely interact with immune cells — through direct contact or soluble mediators — and lead to further elevation of cytokines [22].
2.3. Effects of Cytokines on the Coagulation Cascade
Tissue factor (TF) is the initiator for the extrinsic coagulation cascade and is expressed on a variety of cells at different levels. IL-6 and TNF-α have been shown to stimulate the secretion of a soluble and highly procoagulant isoform of TF from ECs [23]. Moreover, IL-1 and TNF-α can rapidly upregulate TF expression on circulating monocytes, which comprise a large pool of TF [24]. Conversely, the TF-Factor VIIa complex can couple with protease-activated receptors (PARs) to induce cytokine release from the immune cells and ECs, further fueling the CRS [25]. On the other hand, thrombin is involved in the terminal steps of the coagulation cascade and plays a central role in thrombosis propagation. A human study showed that a single bolus of TNF-α was able to induce a rapid conversion from prothrombin to thrombin [26].
In addition to promoting the coagulation cascade, cytokines can also suppress anticoagulant mechanisms. The thrombomodulin/protein C/protein S pathway, an important mechanism to prevent excessive fibrin deposition, can be suppressed by IL-1 and TNF-α. Both cytokines downregulate thrombomodulin activity [27], as well as decrease protein C activity by reducing endothelial protein C receptor (EPCR) expression or stimulating its exocytosis [13,28].
2.4. Effects of Cytokines on the Fibrinolytic System
Fibrinolysis is mediated by plasmin, an enzyme converted from plasminogen by tissue plasminogen activators and urokinase plasminogen activators (tPA and uPA). A delicate balance between the coagulation and fibrinolytic system maintains the homeostasis of vasculature. Cytokines exerting anti-fibrinolytic effects can tilt the system toward a procoagulant state. In vitro, IL-1, TNF-α, and IFN-γ can stimulate ECs to produce plasminogen activator inhibitor type I (PAI-1), the main inhibitor of tPA, resulting in less plasmin production [29,30]. Interestingly, the secretions of tPA and uPA are also parallelly increased with IL-1 and TNF-α [31,32], although the majority of the secreted form have low affinity to plasmin [32]. Studies in healthy volunteers provide more direct evidence of cytokines affecting the fibrinolytic system. Administration of inhibitory cytokine IL-10 inhibits activation of the fibrinolytic system during induced endotoxemia [33]. On the other hand, administration of TNF-α was associated with a transient activation of the fibrinolytic system due to increased secretion of tPA and uPA, followed by a rapid inhibition thereafter, consistent with in vitro results [34].
3. PRESENTATIONS OF CRS-RELATED COAGULOPATHY
Cytokine release syndrome is commonly recognized in primary HLH [35], BiTE therapy [36], CAR T-cell therapy [37], PTCy-based haplo-HSCT [38,39], and various infections, including the severe acute respiratory syndrome (SARS) [40], Middle East respiratory syndrome (MERS) [41], and COVID-19 [2]. However, presentations of coagulopathy differ among different underlying conditions. In this section, presentations of CRS-related coagulopathy in the three best studied scenarios, HLH, CAR T-cell therapy, and COVID-19, are summarized. The pathogenesis of CRS in the three conditions is summarized in Figure 2.
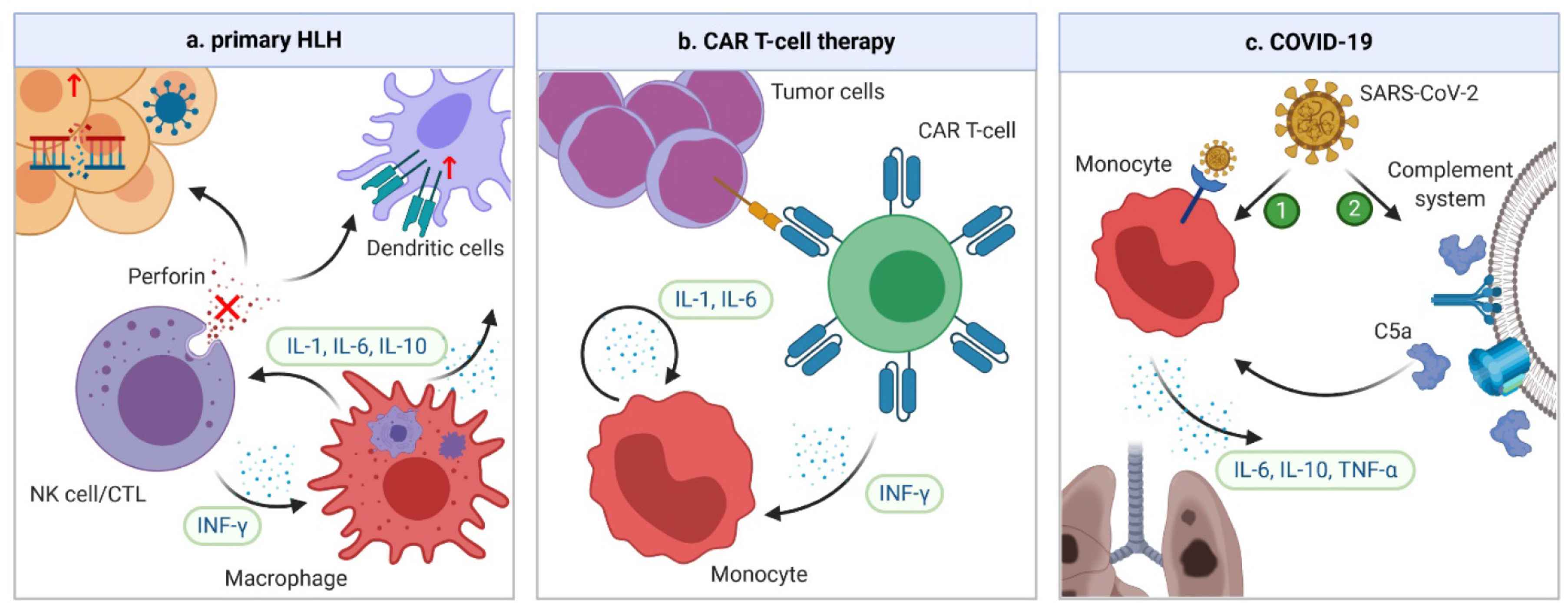
Mechanisms of CRS in HLH, CAR-T cell therapy, and COVID-19. (a) In familial HLH, defects of granule-mediated cytotoxicity (e.g. perforin) in natural killers (NK) cells or cytotoxic lymphocytes (CTLs) lead to impaired killing of diseased human cells. When a trigger, such as a viral infection or genetic mutation occurs, the heightened antigen exposure results in activation of dendritic cells (DCs), whose elimination is further impaired due to defective NK/CTLs. DCs subsequently activate more NK/CTLs, leading to secretion of IFN-γ and activation of macrophages. In turn, macrophages secrete more pro-inflammatory cytokines, leading to a vicious cycle. (b) In CAR T-cell therapy, the CRS is the result of the interplay between CAR T-cells and bystander immune cells. When CAR T-cell and tumor cells are engaged, CAR T-cells are activated and start secreting IFN-γ, which can attract and activate the monocytes. Activated monocytes can secrete key CRS cytokines IL-1 and IL-6, which forms a vicious cycle by activating more monocytes. (c) The pathogenesis of CRS in COVID-19 is unclear. However, it could be secondary to direct infection and activation of immune cells through ACE-2 receptors by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Another possibility is that the complement system can be activated by SARS-CoV-2 infection, which triggers formation of C5a anaphylatoxin. The latter can directly activate neutrophils and monocytes, causing CRS. ACE, angiotensin-converting enzyme.
3.1. Coagulopathy in HLH
Hemophagocytic lymphohistocytosis is a syndrome of fulminant inflammation and tissue destruction caused by unregulated activation of macrophages. Being either familial or secondary to inflammatory conditions, it is characterized by macrophages engulfing different components of blood cells with excessive production of cytokines. Familial or primary HLH is a congenital immune deficiency due to genetic mutations affecting granule-mediated cytotoxicity in natural killers (NK) cells or cytotoxic lymphocytes (CTLs) [42] (Figure 2a). On the other hand, secondary HLH is associated with a variety of inflammatory conditions, such as rheumatologic disorders, malignancies and infection, although the exact mechanisms of secondary HLH remain unclear. Accumulating evidence has suggested a significant overlap between HLH and severe CRS in terms of laboratory findings, cytokine profiles, and clinical presentations [43,44]. In fact, HLH can have the same underlying pathophysiology as severe CRS [44]. The diagnosis of HLH is based on eight criteria: fever, splenomegaly, cytopenia in at least two cell lines, hypertriglyceridemia and/or hypofibrinogenemia, hemophagocytosis, low or absent NK cell activity, hyperferritinemia, and high soluble IL-2 receptor levels. Indeed, these findings are also commonly encountered in severe CRS. The cytokine profile varies slightly with different etiologies of HLH [45], but levels of IFN-γ, IL-6, and IL-10 are universally elevated [35,46,47]. Among them, IFN-γ plays a central role [48]. Coagulopathy is common in patients with HLH. Laboratory findings are consistent with severe consumptive coagulopathy. For example, in a series of 117 patients with HLH due to mixed causes, all patients had thrombocytopenia, 41% had prothrombin time (PT) less than 50% of the normal value, 68% had hypofibrinogenemia, and 50% had disseminated intravascular coagulation (DIC) [49]. In another series of 23 patients with therapy-refractory HLH, 16 (69.6%) had clinical signs of thrombotic microangiopathy (TMA) [50]. Clinically, the coagulopathy predominantly manifests as bleeding, likely due to severe thrombocytopenia and DIC [51]. In a study of critically ill patients with HLH, 22% suffered from severe bleeding and 4.3% died from serious bleeding complications [49].
3.2. Coagulopathy in Anti-CD19 CAR T-cell Therapy
Chimeric antigen receptors are engineered receptors consisting of an ectodomain that can recognize tumor antigen, and an endodomain that can transmit the signal to activate T-cells [52]. Anti-CD19 CAR T-cell therapy has revolutionized the treatment of relapsed/refractory B-cell malignancies with unprecedented durable remissions. CRS, the most common serious adverse effect of CAR T-cell therapy, occurs in about one-half of the patients, and 10–20% experience severe CRS [53,54]. Triggered by activation of CAR T and bystander immune cells, CRS is characterized by significant elevation of CAR T-cell-derived IFN-γ, and monocyte-derived IL-1 and IL-6 [55,56] (Figure 2b). Other cytokines, such as IL-2, IL-8, IL-10, IL-15, MCP-1, and TNF-α are also elevated [4]. Coagulopathy is frequently encountered in CAR T-cell therapy; laboratory findings of mild to moderate consumptive coagulopathy are common. In a study of 100 patients receiving anti-CD19 CAR T-cell therapy, 50% had elevated
3.3. Coagulopathy in COVID-19
COVID-19 is caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Emerging in Wuhan, China, in late 2019 and subsequently spreading worldwide, the infection has a mortality rate as high as 30% in critically ill patients [61]. Since the early spread of COVID-19, high levels of cytokines have been identified in critically ill patients. The levels of IL-2R, IL-6, IL-10, and TNF-α were significantly higher in severe than in non-severe cases or healthy controls [62–64]. In addition, the levels of IL-1, IL-4, IL-7, IL-8, IL-9, granulocyte colony stimulating factor (G-CSF), MCP-1, GM-CSF, and IFN-γ were also mildly elevated in patients with COVID-19 compared with healthy controls [62]. However, the number of IFN-γ-expressing CD4+ T cells was significantly decreased in severe cases [63]; in addition, higher IL-6/IFN-γ ratio was associated with more severe diseases [65], indicating a relatively low IFN-γ level in these cases. Furthermore, the severity of CRS is correlated with survival. In one study, patients who died from COVID-19 had significantly higher IL-6 levels than those who survived [66]. The mechanisms of CRS in COVID-19 are poorly understood. However, given the similarity between SARS-CoV-2 and SARS-CoV, the culprit of SARS, CRS could be due to direct infection and activation of monocytes, macrophages, and dendritic cells, through the angiotensin-converting enzyme 2 (ACE2) receptors on immune cells [67]. Another possible mechanism is activation of the complement system by viral infection, with resultant generation of C5a anaphylatoxin, which can further activate neutrophils and monocytes to cause CRS [68] (Figure 2c).
Common laboratory findings include elevated
3.4. The Many Faces of CRS-related Coagulopathy
Despite CRS being a key feature in all three scenarios and likely a crucial contributor to the coagulopathy, differences exist in laboratory findings and clinical manifestations (Table 1). However, one unifying feature is the development of microvascular thrombosis, although the severity varies among different scenarios. Primary HLH and CAR T-cell therapy are associated with more severe TMA with DIC, while COVID-19 is rarely associated with DIC. This is possibly related to the different severities of CRS. For example, the peak value of IFN-γ, a key proinflammatory cytokine in CRS, is significantly higher in HLH (median 901 pg/mL [35]) and CAR T (1000 pg/mL [80]) than even in the most severe cases of COVID-19 (10 pg/mL [62]). Another universally elevated cytokine in CRS, IL-6, was also higher in HLH (median peak 64 pg/mL [35]) and CAR T (10,000 pg/mL [80]) than severe cases of COVID-19 (40 pg/mL [63]).
| Incidence of CRS | Incidence of coagulopathy | Cytokine profile | Laboratory abnormalities | Pathological presentation | Clinical presentation | |
|---|---|---|---|---|---|---|
| HLH | Close to 100% | Close to 100% | ↑↑ INF-γ | ↑ |
Consumptive coagulopathy | Increased bleeding |
| ↑ IL-6, IL-10 | ↓ Fibrinogen, platelets | Diffuse TMA | ||||
| DIC common | ||||||
| CAR T-cell therapy | Around 50% | Around 50% | ↑↑ IL-6, IL-1 | ↑ |
Consumptive coagulopathy | Increased VTE |
| ↑ INF-γ, IL-2, IL-8, IL-10, IL-15, MCP-1, TNF-α | ↓ Fibrinogen, platelets | TMA in severe disease | ||||
| DIC in severe disease | ||||||
| COVID-19 | Common in severe cases | Prevalence of VTE: 14.1% | ↑↑ IL-2R, IL-6, IL-10, TNF-α | ↑ |
No consumptive coagulopathy | Increased VTE |
| ↑ IL-1, IL-4, IL-7, IL-8, IL-9, G(M)-CSF, MCP-1 | ↔ Platelets, PT, aPTT | Microvascular thrombosis in severe disease | ||||
| ↔ INF-γ | DIC rare |
↔, unchanged. FDP, fibrinogen degradation product.
Epidemiology and presentations of coagulopathy in HLH, CAR T-cell therapy, and COVID-19
Another noticeable difference is the CRS profile. HLH is characterized by markedly elevated IFN-γ level; CAR T-cell therapy is known for significantly elevated IL-1 and IL-6 level; while COVID-19 has relatively low levels of both type I [81] and type II interferons [62]. It is generally believed that elevated proinflammatory cytokines are associated with a procoagulant state, as summarized above. However, little evidence exists as how different cytokines could lead to certain presentations of coagulopathy.
Finally, the underlying conditions associated with CRS may also explain the different presentations. Secondary HLH is often triggered by another severe systemic inflammatory condition, such as fulminant infection or cancer, which themselves are known causes of DIC. Moreover, platelets play a key role in preventing bleeding in inflammation, through mechanisms such as angiopoietin-1 mediated stabilization of endothelial cell–cell junction [82] and direct sealing of endothelial gaps by engaging platelet receptors [83]. Given that HLH and severe CRS are associated with more severe thrombocytopenia, the bleeding risk will be elevated. On the other hand, anti-CD19 CAR T cells are used in patients with relapsed/refractory B-cell malignancies, conditions associated with increased thrombotic risk. The preexisting risk of relapsed/refractory B-cell malignancies combined with CRS-related coagulopathy in CAR T-cell therapy likely contribute to a hypercoagulable state with increased incidence of VTE. As for COVID-19, there are several unique mechanisms on top of the CRS that could lead to further increased risk of thrombosis. First, SARS-CoV-2 can directly infect endothelial cells causing endotheliitis and leading to activation of the coagulation cascade [84]. Moreover, a unique phenomenon in sepsis-related coagulopathy called NETosis is also believed to play a major role [85,86]. Neutrophils, as part of the innate immune system, are activated upon pathogen exposure and can expulse their chromatin decorated with antimicrobial agents which can capture and kill pathogens — a process known as neutrophil extracellular traps (NETs) formation or NETosis [87]. Intravascular aggregation of NETs can lead to occlusion of affected vessels [85]. Moreover, NET itself is a strong activator of platelets, leading to aggregation and thrombosis [88].
4. EFFICACY OF CYTOKINE REDUCTION IN THE MANAGEMENT OF COAGULOPATHY
4.1. Cytokine Inhibitors
Monoclonal antibodies against cytokines or their respective receptors have achieved great success in controlling CRS. For example, in HLH where IFN-γ plays the major role, use of the anti-IFN-γ monoclonal antibody emapalumab has shown promising results in reducing the disease severity [89] and correcting coagulation abnormalities [90]. In addition to IFN-γ inhibition, the effectiveness of IL-1 and IL-6 inhibition in HLH are being actively studied [91,92]. In CAR T-cell therapy, the IL-6 receptor inhibitor tocilizumab has become the first-line treatment for CRS [93]. At the same time, coagulopathy could also be reversed with tocilizumab. For example, in a study of 15 patients who developed signs of DIC after receiving anti-CD19 CAR T-cell therapy and were treated with tocilizumab, 12 (80%) were rescued from DIC within 7 days [58]. If tocilizumab and corticosteroids failed to achieve control of CRS, therapies such as the IL-1 receptor inhibitor anakinra and the IL-6 inhibitor siltuximab can be attempted. Anakinra and siltuximab have extra benefit for mitigating central nervous system CRS, by crossing the blood-brain-barrier and directly blocking IL-6, respectively [94]. Their effects on reversing coagulopathy need to be confirmed.
The cytokine profile in COVID-19 is characterized by high levels of IL-6 and TNF-α, with relatively low IFN-γ. Tocilizumab has been trialed in COVID-19 to mitigate CRS. Although early retrospective studies showed some promising results [95,96], a randomized control trial did not find tocilizumab effective for preventing intubation or death in moderately ill patients [97]. Its effects on coagulopathy, however, remain to be established. Studies regarding other cytokine inhibitors, such as the IL-1 receptor inhibitor anakinra, IL-6 inhibitor siltuximab and anti-TNF antibody infliximab, are ongoing [98,99]. Although the evidence is limited, extrapolating the success in reversing coagulopathy in HLH and CAR T should be cautioned, as some unique thrombotic mechanisms in COVID-19, such as the endotheliitis and NETosis, would not be affected by cytokine inhibition.
4.2. Other Therapies Targeting the Cytokine Pathway
4.2.1. Plasma exchange
Plasma exchange is an extracorporeal blood purification technique that replaces the patient’s plasma with another fluid such as allogenic plasma. It helps maintain hemostasis by removing excessive cytokines and correcting coagulation factors at the same time. It is the standard treatment for a subtype of TMA, thrombocytopenic purpura, through mechanisms of replenishing ADAMTS-13 and removing ADAMTS-13 inhibitors [100]. Plasma exchange has also been tried in DIC with some success [101,102]. While plasma exchange is not a standard therapy for CRS in HLH or CAR T-cell therapy, it has been successfully applied in both scenarios [103,104]. Plasma exchange has also been entertained in COVID-19 and halted disease progression was reported [105]. More evidence is needed to establish the benefit of plasma exchange in CRS-related coagulopathy, especially in patients with severe disease. However, routine use of plasma exchange would not be feasible, due to the requirement for central venous access, specific equipment, and high cost.
4.2.2. JAK inhibitors
Janus kinases (JAK) are the primary downstream signaling pathway of many proinflammatory cytokines such as IL-6 and IFN-γ, with noticeable exceptions such as IL-1 and TNF-α [106]. The JAK inhibitors ruxolitinib, baricitinib and tofacitinib have been successfully applied to treat CRS. In a pilot trial of five patients with secondary HLH treated with ruxolitinib, all patients achieved response [107]. The efficacy of JAK inhibition in controlling CRS in CAR T-cell therapy has been proven in a preclinical model [108]. The efficacy of baricitinib in COVID-19 is being actively tested in the Adaptive COVID-19 Treatment Trial 2 (ACTT-2) trial. Of note, JAK inhibitors have the advantage over cytokine inhibitors due to their oral administration and capacity to inhibit multiple cytokine pathways at the same time. Moreover, in COVID-19, JAK inhibitors have additional anti-viral effects by blocking SARS-CoV-2 entry [109]. However, there has been concern over an increased thromboembolism risk associated with JAK inhibitors in rheumatoid arthritis, based on data from post-marketing surveillance databases, although the events are too few to be certain [110]. Therefore, the effects of JAK inhibitors on cytokine-related coagulopathy need to be carefully studied.
4.2.3. Complement system blockade
The complement system is an innate immune system activated by pathogens such as the SARS-CoV-2. Its activation can directly lead to thrombosis due to endothelial damage. Moreover, complement activation could be upstream of CRS in COVID-19 [68]. Therefore, the use of anti-C5 antibody eculizumab is being tested; preliminary results showed a decrease in cytokine levels [111]. However, its application in the non-COVID-19 setting will likely be limited, due to lack of complement activation in other scenarios.
4.2.4. Corticosteroids
Corticosteroids remain as one of the first-line therapies for HLH. In CAR T-cell therapy, corticosteroids are usually reserved as the last resort for life-threatening CRS, due to concerns of CAR T-cell suppression. The use of corticosteroids in COVID-19 was initially discouraged due to worse outcomes when used in SARS and MERS [112]. However, a prospective study found that hospitalized patients had improved survival when dexamethasone was used [113], likely due to more severe CRS in COVID-19 than SARS and MERS. Corticosteroids have become the standard treatment for patients with COVID-19 requiring supplemental oxygen. The effects of corticosteroids on coagulopathy need to be studied in the future.
4.2.5. Bruton’s tyrosine kinase inhibitor
The Bruton’s tyrosine kinase inhibitor ibrutinib has been shown to also inhibit the IL-2-induced tyrosine kinase, resulting in reduction of cytokine production [114]. In a mouse model, ibrutinib reduced the severity of CRS mediated by CAR T-cell therapy [115]. In a retrospective study of patients with relapsed or refractory chronic lymphocytic leukemia receiving CAR T-cell therapy with or without ibrutinib, the use of ibrutinib was associated with lower CRS severity [116].
5. CONCLUDING REMARKS
In summary, there has been ample evidence to support a direct and causative association between proinflammatory cytokines and coagulopathy. Although the cytokine profile and presentations of coagulopathy differ in the various causes of CRS, similar management methods could be applied. The story behind CRS is an excellent example of how the progress of medical knowledge in one field can facilitate understandings in other fields. If it were not for the extensive research on sepsis-related coagulopathy and CAR T-related CRS, it would be inconceivable to quickly recognize the important crosstalk between cytokine and coagulopathy in COVID-19. Despite broad application of cytokine inhibitors, further studies are needed to confirm their efficacy in reversing coagulopathy.
CONFLICTS OF INTEREST
The authors declare they have no conflicts of interest.
AUTHORS’ CONTRIBUTION
JW and JD wrote the manuscript. JW designed the figures.
FUNDING
No financial support was provided.
REFERENCES
Cite this article
TY - JOUR AU - Jiasheng Wang AU - John Doran PY - 2021 DA - 2021/01/28 TI - The Many Faces of Cytokine Release Syndrome-Related Coagulopathy JO - Clinical Hematology International SP - 3 EP - 12 VL - 3 IS - 1 SN - 2590-0048 UR - https://doi.org/10.2991/chi.k.210117.001 DO - 10.2991/chi.k.210117.001 ID - Wang2021 ER -