
Protocol of the SPARTE Study: A Strategy for Preventing Cardiovascular and Renal Events based on ARTErial Stiffness
 , Gilles Chatellier1, 2, 3, Michel Azizi1, 2, 4, 5, David Calvet6, 7, Gabriel Choukroun8, 9, Nicolas Danchin1, 2, 10, Pascal Delsart11, , Philippe Gosse12, , Gerard London13, Jean-Jacques Mourad14, Bruno Pannier13, Helena Pereira3, Dominique Stephan15, 16, Pierre Boutouyrie1, 2, 17, On Behalf of SPARTE Investigators†
, Gilles Chatellier1, 2, 3, Michel Azizi1, 2, 4, 5, David Calvet6, 7, Gabriel Choukroun8, 9, Nicolas Danchin1, 2, 10, Pascal Delsart11, , Philippe Gosse12, , Gerard London13, Jean-Jacques Mourad14, Bruno Pannier13, Helena Pereira3, Dominique Stephan15, 16, Pierre Boutouyrie1, 2, 17, On Behalf of SPARTE Investigators†The list of all centers is given in Supplementary file.
- DOI
- 10.2991/artres.k.200711.001How to use a DOI?
- Keywords
- Antihypertensive drugs; arterial stiffness; cardiovascular events; clinical trial; hypertension
- Abstract
Whether arterial stiffness is a surrogate end-point for cardiovascular and renal disease has never been directly demonstrated by a controlled clinical trial. Our main hypothesis is a better prevention of outcomes in high risk hypertensives with PWV normalization driven strategy than with usual blood pressure driven therapeutic strategy based on European Society of Hypertension–European Society of Cardiology (ESH–ESC) guidelines. The strategy for preventing cardiovascular and renal events based on arterial stiffness study is a multicenter open-label randomized controlled trial with blinded endpoint evaluation comparing a therapeutic strategy targeting the normalisation of Pulse Wave Velocity (PWV group) versus a classical therapeutic strategy only implementing the ESH–ESC Guidelines (conventional group), for reducing cardiovascular and renal events. Patients with primary hypertension, aged 55–75 years, and at medium-to-very high cardiovascular risk will be included and followed-up for 4 years. In the PWV group, treatment will be adjusted to carotid-femoral PWV measured every 6 months. In the conventional group, PWV will be measured at baseline and every 2 years, but its value will be blinded to the investigator in charge of the patient. In the PWV group, the therapeutic strategy will preferably use a combination of Angiotensin-converting Enzyme Inhibitor (ACEI) [or Angiotensin Receptor Blockers (ARB)] and calcium channel blockers, as well as maximal recommended doses of ACEIs and ARBs. The primary combined endpoint includes stroke and coronary events (myocardial infarction, angioplasty, bypass), fatal or not, peripheral artery disease (angioplasty, bypass, amputation), hospitalization for heart failure, aortic dissection, chronic kidney disease (doubling of creatinine, dialysis), and sudden death. Twenty-five research centers will include a total of 1500 patients, in order to show a 20% reduction in the primary combined endpoint - the incidence of which is estimated at 10% per year - in the PWV group compared to the conventional group.
- Copyright
- © 2020 Association for Research into Arterial Structure and Physiology. Publishing services by Atlantis Press International B.V.
- Open Access
- This is an open access article distributed under the CC BY-NC 4.0 license (http://creativecommons.org/licenses/by-nc/4.0/).
1. INTRODUCTION
The treatment of hypertension is associated with the treatment of other Cardiovascular (CV) risk factors, in a “global risk” approach. Hypertension Mediated Organ Damage (HMOD) increases CV risk, independently of Blood Pressure (BP) level. Although the treatment of hypertension aims at lowering BP to better reduce target organ damage and ultimately reduce CV and renal complications, very few studies have shown that a strategy aimed at reducing HMOD translates to reduction of CV and renal complications beyond BP reduction. Indeed, we know that for similar reduction in BP, the regression of HMOD differs markedly. This has been well demonstrated for the treatment-induced regression of Left Ventricular Hypertrophy (LVH) measured by either Electrocardiogram (ECG) or echocardiography in the Losartan intervention for endpoint reduction in hypertension study (LIFE) [1–3], thus qualifying them as surrogate endpoints. However, results are less consistent regarding the translation of a treatment-induced reduction in urinary albumin excretion into a reduced incidence of CV events and slower progression of renal disease [4–6].
An exaggerated arterial stiffness, characterized by an elevated Carotid-femoral Pulse Wave Velocity (cfPWV > 10 m/s) [7] can be considered as HMOD, here the organ being the aorta. The corrected 10 m/s threshold, that is also included in the 2013 European Society of Hypertension (ESH)/European Society of Cardiology (ESC) Guidelines for the management of hypertension [8], corresponds to the previous threshold of 12 m/s that figures in the 2007 ESH–ESC Guidelines [9], adjusted for carotid-femoral distance (coefficient 0.8) according to the 2012 international consensus [7]. However, aortic stiffness cannot qualify yet as such because no controlled study has shown until now that the reduction in arterial stiffness translates to reduction of CV and renal complication independently of BP reduction in hypertensive patients.
In order to be considered as a surrogate endpoint of CV events, a biomarker should satisfy several criteria, such as proof of concept, prospective validation, incremental value, clinical utility, clinical outcomes, cost-effectiveness, ease of use, methodological consensus, and reference values [10]. The repeated demonstration of the predictive value of arterial stiffness for CV events led to its inclusion in the 2013 and 2018 ESH/ESC Guidelines for the management of Hypertension [8,11], as HMOD. A position paper from the European Society of Cardiology - working group on peripheral circulation [12] scrutinized the role of peripheral (i.e. not related to coronary circulation) noninvasive vascular biomarkers for primary and secondary CV disease prevention. cfPWV was one of the few biomarkers that fulfilled most of the criteria and, therefore, was close to being considered a clinical surrogate endpoint [12]. Finally, a recent call to action of the Lancet Commission on Hypertension [13] addressed the global burden of raised BP through a life-course strategy based on the quantification of early vascular ageing, an equivalent of arterial stiffness [14]. We thus set up the SPARTE trial as a Strategy for Preventing Cardiovascular and Renal Events based on ARTErial Stiffness. A detailed rational has been previously published [15].
In the SPARTE study, we hypothesized that a therapeutic strategy including the normalisation of arterial stiffness in addition to the implementation of international guidelines for normalisation of BP would reduce more CV and renal events compared to the unique implementation of the international guidelines for the management of hypertension. The objective of the present paper is to present the protocol in detail, to report the dates of the study and the inclusion dynamics, and discuss some feasibility elements.
Of note, the protocol has been submitted to ethical committee and funding institutions in 2012. Thus, 2007 ESH–ESC Guidelines for the management of hypertension [16] applied at that time. However, the references which appear in the present document in order to support the rational of the study, particularly those related to pharmacological treatment, have been updated.
2. MATERIALS AND METHODS
Strategy for preventing cardiovascular and renal events based on arterial stiffness is a multicenter study including 25 French clinical centers, among which 12 are Excellence Centers labelled by the ESH. It is investigator initiated and driven, funded by the French Ministry of Research (PHRC 2011- K110102 / N°ID RCB: 2012-A00023-40) and Fondation de Recherche sur l’Hypertension Arterielle (FRHTA). A list of the study sites is provided in Table S1.
2.1. Study Design
The trial is defined by the protocol NCT02617238 (http://www.clinicaltrials.gov/) as a multicentre randomised two parallel groups study using a prospective, randomised, open, blinded end-point design, aiming at comparing the efficacy of a therapeutic strategy targeting the normalisation of arterial stiffness for reducing CV and renal events (PWV group), in comparison with a classical therapeutic strategy implementing the ESH–ESC Guidelines [16] (conventional group), in patients with primary hypertension and medium-to-very high CV risk (Figure 1).
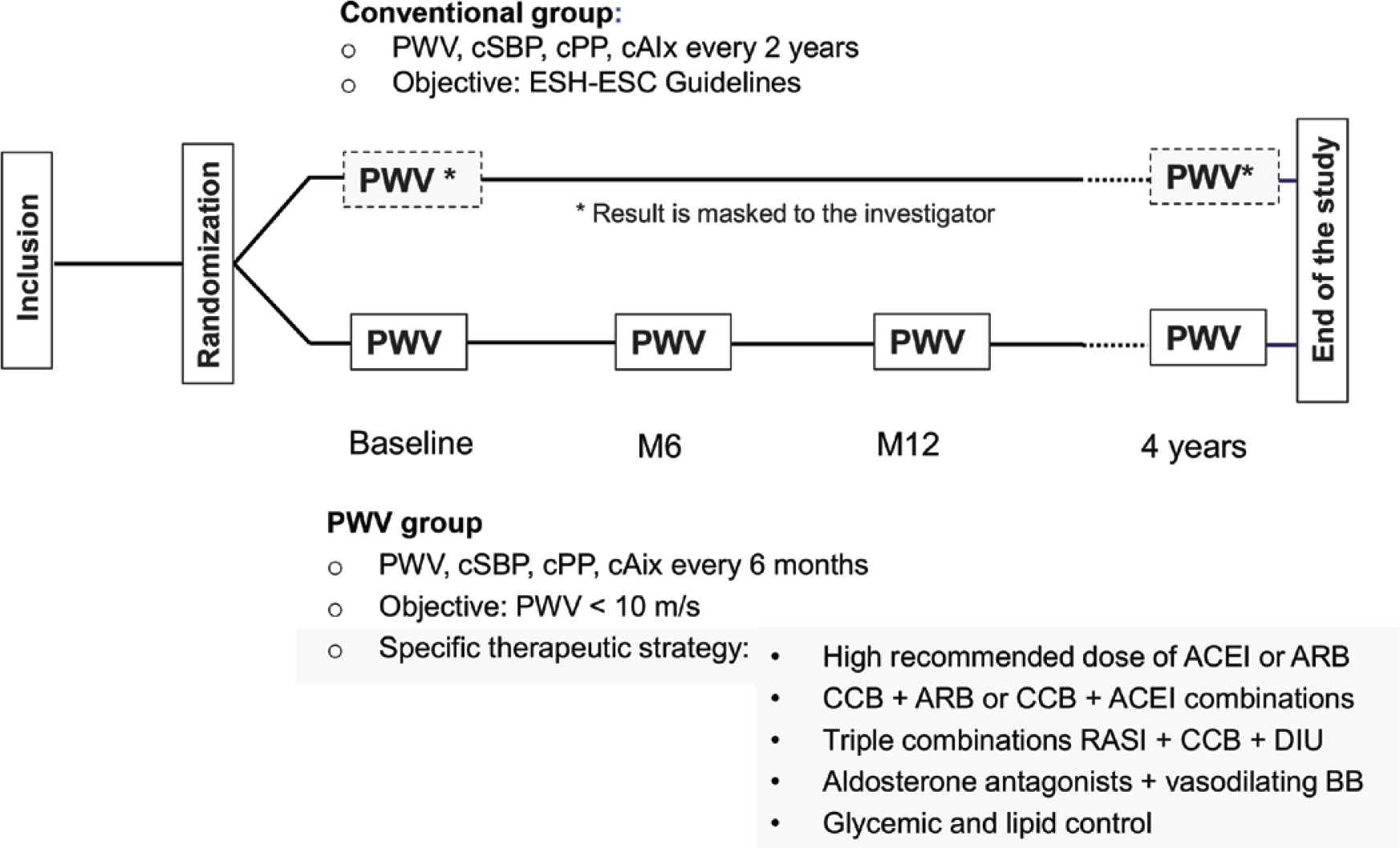
Experimental design of the SPARTE study. The SPARTE study is a 4 years multicentre randomised two parallel groups study using a PROBE design, aiming at comparing the efficacy of a therapeutic strategy targeting the normalisation of arterial stiffness for reducing cardiovascular and renal events (PWV group), in comparison with a classical therapeutic strategy implementing the ESH–ESC Guidelines (conventional group), in patients with primary hypertension and medium-to-very high CV risk.
Since treatment of hypertension is adjusted on the repeated measures of PWV in the PWV group, SPARTE study is truly a “target driven, long-term intensified intervention trial”.
- •
Target driven study: The target is not BP such as in ACCORD [17], CARDIO-SiS [18], JATOS [19] or UKPDS [20], but arterial stiffness. The target value of PWV is defined as 10 m/s, recommended by the 2012 “Expert consensus document on the measurement of aortic stiffness in daily practice using carotid-femoral pulse wave velocity” [7]. The value of 10 m/s is consistent with the median values of the 55–75 years categories, observed in the Reference Value - Arterial Stiffness Collaboration Study [21].
- •
- •
Intensified intervention trial: A number of pharmacological studies have shown that such a BP-independent lowering of arterial stiffness is best obtain using blockers of the renin–angiotensin–aldosterone system, including high recommended doses of Angiotensin-converting Enzyme Inhibitors (ACEI) [22,26] and Angiotensin Receptor Blockers (ARB) [24,27] and when necessary mineralocorticoid receptor blockers [28]. The therapeutic strategy is detailed below.
2.2. Sample Selection
The coordinating center in Paris supplied the study documentation to all participating centres. Study documentation has included a description of the protocol and of the expected tasks, and the files to be signed in relation to study participation agreement.
2.3. Screening and Randomization
Around 60 subjects were to be enrolled in each center upon verifying the study eligibility criteria listed in Table 1.
| Inclusion criteria |
|
| Exclusion criteria |
Cannot be included:
|
ISH, isolated systolic hypertension; LVMI, left ventricular mass index.
SPARTE inclusion and exclusion criteria for enrolment
Following a parallel group study design, eligible patients are randomized one-to-one in the treatment arms. The randomization list, created by the coordination center before the start of inclusions, uses a randomized block design, and is stratified by center and categories of CV risk. The randomization is centralized and made available to centres through the web-based software CleanWeb (Telemedicine Technologies, http://www.tentelemed.com/la-solution-cleanweb/). At randomization, the clinician obtains the patient allocation group after having recorded the patient characteristics required by the algorithm.
In each centre, one or more investigators are in charge of the screening. After the initial visit and signature of the informed consent, checking inclusion and exclusion criteria, the patients are randomised. At inclusion visit, a clinical examination is performed and the following elements will be recorded: Height, weight, waist circumference; Systolic Blood Pressure (SBP), Diastolic Blood Pressure (DBP) (three measurements with oscillometric device); Risk factors (lipids, diabetes, hypertension, smoking); CV disease history; Noncardiovascular disease history; HMOD; Biological dosages (not more than 6 months before): creatinine and Estimated Glomerular Filtration Rate (eGFR)-Modification of Diet in Renal Disease (MDRD), blood glucose, total, HDL, LDL Cholesterol (LDL-C), triglycerides, Na, K, albuminuria; Ongoing treatments (pharmacological classes and doses).
Ambulatory Blood Pressure Monitoring (ABPM) is performed immediately before the initial visit. Patients without treatment who have normal BP at ABPM (24 h BP < 130/80 mmHg or day BP < 135/85 mmHg) are not included in the study. In case ABPM is not possible, Home Blood Pressure Monitoring (HBPM) is performed, and patients with normal BP (<135/85 mmHg) are not included.
At baseline, the measurement of the PWV and central BP is done immediately before or after the clinical workup. During the study, in the PWV group, the value of PWV is made immediately available for the investigator. In order to improve the flow of patients during the course of the study, this is the PWV measure closest to the clinical visit that is used for adjusting treatment. In the conventional group, the investigator in charge of the patient is blinded to the value of the PWV. PWV is measured every 6 months for the PWV group only. For the conventional group, PWV is measured at baseline, 24 and 48 months (Figure 1). Whatever the group, in case of early termination of the study for one patient, PWV is measured at the time closest to the event, when possible within 15 days of the event. Central BP and Augmentation Index (AIx) is measured during each PWV measurement sessions, but results will not be available to investigators of the PWV group who adjust treatment on PWV only, and, by definition, to investigators of the conventional group.
2.4. Pulse Wave Velocity and BP Measurements
Carotid-femoral PWV is measured by applanation tonometry using the Sphygmocor device (Atcor Medical, Sydney, Australia) using the foot-to-foot method [7,29]. Briefly, the applanation probe is positioned on the carotid artery, in order to record the carotid pressure waveform [7,29]. Applanation is then performed immediately afterward on the common femoral artery. Pulse transit times from concomitant ECG are calculated using the intersecting tangent automatically calculated by the software. cfPWV is calculated by dividing travelled distance by the difference in transit times. For calculating travelled distance, a coefficient of 0.8 is applied to the direct (carotid-to-femoral) path length according to Van Bortel et al. [7]. At least two PWV measurements are performed. If the difference between the two measurements is more than 0.5 m/s, a third measurement is taken and the median value is retained [7].
Central BP is measured by applanation tonometry using the Sphygmocor device (Atcor Medical, Sydney, Australia) as described previously and recommended [7,29,30]. Briefly, the applanation probe is positioned on the radial artery (right arm), and optimal applanation is obtained using visual inspection and following built-in quality control indices. Radial waveforms are calibrated using brachial SBP and DBP measured before and after applanation (average). The central aortic waveform is calculated by the device software using the generalized transfer function. BP values are derived from the curve. AIx is measured, and AIx at heart rate 75 (AIx@75) is calculated through the software.
Brachial SBP and DBP and Heart Rate (HR) is measured using an adapted cuff with any validated electronic oscillometric device, both at the physician’s office during the outpatient clinic and at the tonometry center, according to ESH–ESC guidelines. Three measurements are performed, and the average of measures 2 and 3 is retained.
Quality control is done before and during the study. All applanation tonometry centers are already experienced with arterial measurements. They have all undergo training both at the Core Lab Facility of Pharmacology Department in Pompidou Hospital and on site. Centers are certified on the basis of five consecutive measurements fulfilling pre-established quality features. All measurements are centrally reviewed by the Core Lab immediately after having being performed. A trained technician, blinded as to the center, period and treatment, checks for quality of tracings and inconsistencies in BP values. In the event of a mismatch, BP values could either be corrected or measured again within 1 week. During the course of the study, arterial measurements are randomly sent to the Core Lab Facility of Pompidou Hospital, checked again, and appropriate measures are to be taken to maintain a high level of quality.
2.5. Duration of the Study
The follow-up study duration is 4 years, during which at least two visits are performed every year for each group (see Table 2 for detail). In the PWV group, one visit every 2 months is performed during the first 6 months in order to adjust treatments to PWV target of less than 10 m/s, and then every 6 months. In the conventional group, visits will occur at least twice a year (Figure 1).
| Period | Selection/Inclusion | Trial | End of study | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Visit | 1 | 2 | 3* | 4 | 5* | 6 | 7* | 8 | 9* | ... | 18 |
| Month | −1 à 0 | 2 | 4* | 6 | 9* | 12 | 16* | 18 | 20* | ... | 48 |
| Visit location | C°HT or PWV centre | C°HT | C°HT | C°HT or PWV centre | C°HT | C°HT or PWV centre | C°HT | C°HT or PWV center | C°HT | ... | C°HT or PWV center |
| Informed consent | X | ... | |||||||||
| Inclusion/exclusion criteria | X | ... | |||||||||
| Randomisation | X | ... | |||||||||
| Medical history | X | ... | |||||||||
| Clinical examination | X | X | X | X | X | X | X | X | ... | X | |
| ABPM or HBPM | X | X | ... | X | |||||||
| PWV measurement | X1 | XPWV | XPWV | XPWV | ... | X1 | |||||
| Laboratory tests | X | X* | X* | X* | X* | X* | X* | X* | X* | X* | |
| Adverse events | X | X | X | X | X | X | X | ... | X | ||
| Medication report | X | X | X | X | X | X | X | X | ... | X | |
| eCRF | X | X* | X* | X | X* | X | X* | X | X | X | |
all visits labelled as (*) are optional, in order to better adjust treatment, with a minimum of two visits per year (every 6 months).
The measurement of cfPWV in the PWV group should be performed before the outpatient clinic, in order to better adjust the treatment. C°HT, outpatient clinic; Centre PWV, PWCV can be measured in another centre than the outpatient clinic; X*, As required by the investigator; PWV measurement: XPWV, every 6 months for the PWV group only; X1, PWV at V1, V10 (24 months) and V18 (48 months) for the conventional group.
Time schedule of enrolment, interventions, assessments and visits of participants
At each of the follow-up visits, the following elements are recorded: Weight, height, waist circumference; Office SBP and DBP; Heart rate; Laboratory tests if required; Smoking; Ongoing treatments, including antihypertensive, lipid lowering, antidiabetic, and antiplatelet drugs; Clinical events: Stroke, coronary events, peripheral artery events, hospitalisation for heart failure, kidney events, and sudden death.
Ambulatory blood pressure monitoring (or HBPM if ABPM is not available) is performed at baseline, after 6 months and at the end of the study. The last visit is similar to the follow-up visits, except that PWV, ABPM (or HBPM if ABPM is not possible) and laboratory tests are systematically performed regardless of the group to which the patient belongs.
2.6. Blinding
Because SPARTE is an open-label study, blinding will apply for the endpoints in both groups (PWV group and conventional group). In the PWV group where PWV measurement is used to adjust the therapeutic strategy, both patients and investigators are aware of PWV values. In the control group, investigators and patients are blinded for PWV because PWV measurement is not used for adapting therapeutic strategy and only serves for comparing groups afterward.
All components of the primary outcomes of the study are adjudicated in a blinded fashion (allocation group and PWV value) by the Endpoint Adjudication Committee (EAC). The Clinical Research Unit (CRU) of Hopital Europeen Georges Pompidou (HEGP) in Paris serves as coordinating center and sends all outcome documentations to each of the three EAC experts (D.C., G.C. and N.D.). In case of discrepancy, a face-to-face meeting or a teleconference is organised by the Paris CRU in order to reach a consensus. Only validated outcomes will enter the final data analysis.
2.7. Interventions
In the conventional group: no specific therapeutic strategy is done other than those included as mandatory in the ESH–ESC guidelines for the management of hypertension. The objective is to bring BP below 140/90 mmHg [16], targeting 130–139 mmHg for SBP and 80–85 mmHg for DBP [16]. These guidelines are followed not only for antihypertensive treatment but also for caring about other risk factors, in addition to other international guidelines.
In the PWV group, the objective is to bring PWV below the target of 10 m/s [7]. For that purpose, antihypertensive treatment is adjusted and CV risk factors corrected until normalisation of PWV. More specifically, in patients with controlled BP, the first goal is to normalise PWV. In patients with high BP, the first goal is to normalise BP, and then to adjust treatment for normalising PWV. Indeed, as discussed below, PWV can be reduced through both acute BP reduction (passive destiffening) and long-term BP reduction (arterial remodeling). In those patients, is it important to take into account the time delay between the normalisation of BP and that of PWV. This delay can reach several months [23]. Of note, the two goals (normalising BP or normalising PWV) could have overlapped since the therapeutic strategy used for normalising BP is also effective for normalising PWV beyond BP.
Therapeutic means to be used in the PWV group are detailed below in Table 3. The therapeutic strategy puts into application the results of several pharmacological studies comparing the efficacy of various pharmacological classes for lowering arterial stiffness independently of BP reduction. Several studies unequivocally showed that antihypertensive drugs are able to reduce arterial stiffness and/or wave reflections independently of the reduction in brachial BP, for instance after acute calcium channel blocker administration [31], after long-term ACE inhibition by perindopril [22] or trandolapril [26], and after long-term angiotensin-receptor blockade by valsartan [30] or olmesartan [25]. In addition, the antialdosterone drug spironolactone is able to reduce arterial stiffness beyond BP reduction [28]. Several reviews have concluded that Renin–Angiotensin System (RAS) blockers and Calcium Channel Blockers (CCB) are more potent than betablockers and diuretics for reducing arterial stiffness beyond BP reduction [24,32].
| There are two possibilities |
| I. BP is controlled at entry |
| see II.b.2 |
| II. BP is not controlled at entry |
| II.a. First step |
The first objective is to target a BP < 140 and < 90 mmHg, in the range 130–140 mmHg SBP and 80–85 mmHg, Combination therapy for all:
|
| II.b. Second step |
| II.b.1. Either BP remains not controlled |
| Use a triple combination: DIU + ACEI (or ARB) + CCB |
And then, if BP is still not controlled or side effects occur, as third step:
|
| II.b.2. Or BP is controlled but PWV is not reduced/normalised |
|
| Anytime, if BP is too low, go back one or more steps |
HCTZ, hydrochlorothiazide.
Therapeutic strategy in the PWV group
Combinations therapy using a RAS blocker (ACEI or ARB) and a CCB is recommended as first step, according to the 2007 ESH–ESC Guidelines for the management of hypertension [16], for a more effective control of BP. Regarding their effects on the arterial wall, the evidence originates mainly from studies on central BP. The following combinations have demonstrated their effectiveness for lowering central BP in the CAFE study [33] using the perindopril/amlodipine combination, the EXPLOR study using the valsartan/amlodipine combination [30], and the Japan-combined treatment with olmesartan and a calcium channel blocker versus olmesartan and diuretics randomized efficacy study (J-CORE) using the olmesartan/azelnidipine combination [34]. In the J-CORE study [34], the reduction in PWV was twice larger after olmesartan/azelnidipine than after olmesartan/hydrochlorothiazide. When a diuretic is indicated, indapamide should be preferred. This is based on the results of the preterax in regression of arterial stiffness in a controlled double-blind study (REASON), testing the effectiveness of the perindopril/indapamide combination on central SBP and PWV, compared to atenolol alone [35,36].
Beta-blockers are used as 4th line therapy, unless compelling indication. Each time betablockers are necessitated, a vasodilating one is used if no elective indication for a non-vasodilating betablocker exists (heart failure, atrial fibrillation, etc…). Vasodilating beta-blockers, such as nebivolol [37] or celiprolol [38,39], are preferred for their neutral or positive effect on central BP. Non vasodilating beta-blockers, such as atenolol should be avoided, since this pharmacological class has shown deleterious effect on central BP, arterial stiffness [32,35], small artery remodeling [40], and left ventricular hypertrophy [41,42]. However, vasodilating beta-blockers are indicated only as 4th step, since a pro-fibrotic effect on the arterial wall cannot be excluded on the long term [39].
Spironolactone is used as an alternative to BB as 4th line therapy.
Anytime, in case of poor treatment tolerance (for instance if BP is too low, or adverse reaction, or orthostatic hypotension), a step down is authorized.
Other CV risk factors are corrected according to international guidelines, in particular:
- •
smoking cessation, associated if necessary, by support for smoking cessation
- •
limited salt intake (NaCl) up to 6 g/day
- •
weight reduction in overweight patients, to maintain BMI (body mass index) below 25 kg/m2, or, failing that, to achieve a 10% reduction of the initial weight
- •
regular physical activity, tailored to the patient’s clinical condition, at least about 30 min three times a week
- •
limited alcohol intake to less than three glasses of wine or equivalent per day in men and two glasses of wine or equivalent per day for women;
- •
diet rich in vegetables, fruits and low in saturated fat (animal fat).
Oral antidiabetic agents, lipid lowering agents and antiplatelet agents are used as recommended by international guidelines.
2.8. Outcomes
2.8.1. Primary endpoint
The primary endpoint is a combined endpoint including first CV events, fatal or not: Stroke, coronary event [Myocardial Infarction (MI), angioplasty, bypass], Peripheral Artery Disease (PAD) (angioplasty, bypass, amputation), hospitalization for heart failure, aortic dissection, Chronic Kidney Disease (CKD) (doubling of creatinine, dialysis), sudden death. Patients who have presented an event are followed according to the methods recommended by learned societies and/or the French Ministry of Health guidelines. Recurring events are recorded and validated by an independent committee. On purpose, are not included Transient Ischemic Attack (TIA) and new onset of atrial fibrillation.
2.8.2. Secondary endpoints
- •
“Restricted” combined endpoint, including fatal CV events and non-fatal MI and stroke.
- •
All individual components included in the combined endpoint are analysed separately: stroke, coronary event (MI, angioplasty, bypass), PAD (angioplasty, bypass, amputation), hospitalization for heart failure, aortic dissection, CKD (doubling of creatinine, dialysis), sudden death.
- •
Percentage of normalization of the PWV at the end of study in each group.
- •
Values of PWV, central BP [SBP and Pulse Pressure (PP)] and AIx at the end of study.
- •
Time-course changes in brachial BP (SBP, DBP, mean blood pressure (MBP) and PP) measured at office, at home, and during PWV measurement sessions; in central SBP, PP and AIx; in PWV; in Ambulatory BP (24 h, day, and night SBP and DBP); and in biological parameters (particularly GFR estimated by MDRD).
- •
- •
Time-course changes in treatments, in terms of pharmacological classes and doses (estimated as low, medium and high).
All clinical events are judged by an independent EAC blinded to the group. Clinical endpoints are prespecified with a detailed description in order to help physicians to report endpoints and to allow experts of the EAC to confirm or rule out diagnosis. The first qualifying event is used for morbidity and mortality analysis. Each of the events of the combined endpoint are validated by the EAC. The values of PWV, central BP, brachial BP, ABPM and HBPM are established as set out in the Protocol. It is the same for biological values, European scores (SCORE) and Framingham risk scores.
2.9. Sample Size and Power Consideration
The sample size calculation has been performed using nQuery Advisor® 5.0 (Statistical Solutions, Saugus, MA, USA). A proportion test was used as an approximate estimation for the sample size calculation (two-sided Z-test with unpooled variance). The sample size of the study has been calculated from the main criteria (combined endpoint): stroke + coronary events (MI, angioplasty, bypass) + PAD (angioplasty, bypass, amputation) + hospitalization from congestive heart failure + aortic dissection + doubling plasma creatinine + end stage renal disease + sudden death.
Considering a yearly incidence of the combined endpoint of 10% per year, a 20% risk reduction by the therapeutic strategy targeting PWV, a 4-year follow-up period and an alpha risk of 5%, a sample size of 1500 patients per group gave a power high enough for analysing both the combined endpoint and the “restricted” combined endpoint including fatal and non-fatal MI and strokes. A detailed explanation is given below.
According to the Cardio-Sis [18], ACCORD [17], and STENO [45,46] studies, one can reasonably estimate an incidence of composite events in the SPARTE study of about 10% per year, and their reduction through targeted strategy on PWV 20%. On this basis, we calculated the following estimates after a 4-year follow-up period. A sample size of 1500 patients per group provides a high power (99%) for analysing the composite primary endpoint, and an acceptable power (70%) for the “restricted” endpoint (fatal and non-fatal stroke or MI), thus allowing performing a subgroup analysis with a reasonable power (Table 4).
| N patients | N events control | N events intervention | Difference | Power | Alpha |
|---|---|---|---|---|---|
| 250 | 100 | 80 | 20 | 0.46 | 0.05 |
| 500 | 200 | 160 | 40 | 0.75 | 0.05 |
| 750 | 300 | 240 | 60 | 0.9 | 0.05 |
| 1000 | 400 | 320 | 80 | 0.96 | 0.05 |
| 1250 | 500 | 400 | 100 | 0.98 | 0.05 |
| 1500 | 600 | 480 | 120 | 0.99 | 0.05 |
| Prediction of fatal CV events and non-fatal MI and stroke | |||||
| 250 | 40 | 32 | 8 | 0.17 | 0.05 |
| 500 | 80 | 64 | 16 | 0.30 | 0.05 |
| 750 | 120 | 96 | 24 | 0.42 | 0.05 |
| 1000 | 160 | 128 | 32 | 0.53 | 0.05 |
| 1250 | 200 | 160 | 40 | 0.62 | 0.05 |
| 1500 | 240 | 192 | 48 | 0.70 | 0.05 |
Prediction of primary endpoint
2.10. Data Collection
The study uses an Electronic Case Report Forms [eCRF (CleanWeb, Telemedicine Technologies)]. All information required by the protocol is recorded on the eCRF. The data collected and stored outside eCRF (source data) is transcribed in the notebooks accurately and completely. Any handwritten note on the source data is dated, marked by initials and signed. An explanation is provided for each missing data. Erroneous data found on paper documents is clearly crossed and new data is copied next to the barred information, accompanied by the initials, date, and optionally a justification by the investigator or an authorized person who has made the correction.
2.11. Data Management
Data management is performed by the CRU of HEGP. A list of the variables of the study size operational data model is developed by the CRU, in connection with the study investigators. This list will allow the development by the CRU of data collection specifications and computerized database (PostgreSQL format) where data is entered after quality control. A data-management plan, developed jointly by the data-manager, the investigator and the senior statistician are implemented. After correction of errors, the database will be frozen for statistical analysis.
2.12. Statistical Analysis
The descriptive analysis is carried out as follows: number of values, mean (standard deviation) or median (25% and 75% percentiles) for the quantitative variables; and numbers (percentages) for categorical variables. A flow diagram (flow chart) will be provided as recommended by the Consort Statement [47] including the numbers of eligible patients randomized, and the numbers of patients who fully comply with the protocol, left the study or are lost to follow-up. The major protocol deviations and study output patterns will be described.
The analysis will be performed according to the intention to treat principle, keeping patients in their randomization group, and including protocol violations. We will perform a “per protocol” sensitivity analysis including only patients who fully complied with the protocol. The estimate of survival (overall and progression-free) will be done through the method of Kaplan and Meier. The primary analysis will focus on the combined primary criterion. All components of the primary outcome will be analysed separately. For clinical criteria, effect sizes will be estimated by the hazard ratio calculated with a Cox proportional hazard model, after verification of the hypothesis of proportionality of hazards. All estimates will be provided with their 95% confidence intervals. For other endpoints (PWV changes during protocol for instance), mixed models with time as within-, and group as between-effect, and meaningful covariates (BP changes for instance) will be used. Significance will be fixed at p < 0.05. Analyses will be performed using the SAS software version 9.4 (SAS Institute Inc., Cary, NC, USA) under the responsibility of Prof. Gilles Chatellier, CRU Coordinator at HEGP.
2.13. Ethical Considerations
The SPARTE study protocol has initially received approval by the Ethics Committee central pulse pressure (CPP) of Ile-de-France XI, on June 14th 2012, that is applicable to all participating centers. This is an investigator generated and driven study and as such is performed in full independence of the study sponsors, i.e. “Assistance Publique-Hôpitaux de Paris, Direction de la Recherche Clinique et du Développement”, and “FRHTA”.
According to the bioethics laws, the investigator has the obligation to inform the patients before their recruitment for clinical research studies, even if these studies are common clinical care. In accordance with the local rules, the information of patients participating to research is ensured with a written document previously validated by the Ethics committee. The investigator from the center offers the patient to participate in the study, orally informs him/her about the modalities of the study and deliver him/her the information note. The investigator mentions the “non-opposition” in the patient’s medical record in case of acceptance to participate. In case of refuse, this information will also be mentioned there (and the patient will not be included). When the study is completed, the participating patient may be informed of the overall results of this research in a manner that is specified in the information document.
According to the French bioethics law, there is no need of informed consent here because the SPARTE protocol is aiming at evaluating usual clinical care, by comparing two therapeutic approaches using therapeutic means and drugs already recommended by national or international guidelines, without added risk and with few constraints. Indeed, the algorithm for intensifying antihypertensive treatment in the PWV group is in accordance with the French guidelines, issued by the French Ministry of Health, Haute Autorité de Santé [48]. An informed consent would have been required if, for instance, the protocol would have included a novel drug.
2.14. Progress of the Study
The first patient was included on July 26, 2013. The last patient-last visit occurred on January 26, 2020. The inclusion rate and consequently the total number of patients were lower than expected because of competing protocols in several centers. The protocol is currently being replicated in Poland (Prof. Krzysztof Narkiewicz, Gdansk University) and in Portugal (Prof. Pedro Cunha, Guimaraes-Minho University).
CONFLICTS OF INTEREST
S.L. has received honoraria as lecturer for Menarini, Sanofi and Servier, and Axelife, Omron and Withings. G.C. none. M.A. has received research grants from the French Ministry of Health, Quantum genomics and European H2020 program; has received grant support and non-financial support from ReCor Medical and Idorsia; and has received personal fees from CVRx. D.C. none. G.C. has received honoraria as lecturer, participant to board of experts, and travel grants from Astellas, Sanofi, VIfor Pharma, and Amgen. N.D, P.D., P.G. and G.L. none. J.J.M. has received honoraria for consultancy or lectures for Mylan, Pfizer and Servier. B.P. and H.P. none. D.S. has received honoraria as lecturer for Servier. P.B. currently serves as President of ARTERY sponsored by an unconditional grant by SERVIER.
AUTHORS’ CONTRIBUTION
SL, GC, MA, GL, JJM, BP and PB contributed to the conception and design of the study. SL, GC and PB drafted and wrote the protocol in accordance to the coauthors’ contributions. All authors contributed to the writing and the reviewing of this article, and approved the final draft of the protocol.
FUNDING
French Ministry of Research (PHRC 2011- K110102 / N°ID RCB: 2012-A00023-40) and Fondation de Recherche sur l’Hypertension Arterielle (FRHTA). Centers received funding from the
ACKNOWLEDGMENTS
The names of the collaborating centers and investigators of the SPARTE Study are listed in Supplementary file. Chantal Andrieux (CRU of Georges Pompidou Hospital) acted as clinical study manager from 2013 to 2019.
ABBREVIATIONS
- ABPM,
ambulatory blood pressure monitoring;
- ACEI,
angiotensin converting enzyme inhibitor;
- AP-HP,
Assistance Publique – Hôpitaux de Paris;
- ARB,
angiotensin receptor blockers;
- BP,
blood pressure;
- CCB,
calcium channel blockers;
- CKD,
chronic kidney disease;
- CRF,
case report form;
- CRU,
clinical research unit;
- CV,
cardiovascular;
- DBP,
diastolic BP;
- EAC,
Endpoint Adjudication Committee;
- ECG,
electrocardiogram;
- eGFR,
estimated glomerular filtration rate;
- ESC,
European Society of Cardiology;
- ESH,
European Society of Hypertension;
- HBPM,
home blood pressure monitoring;
- HEGP,
Hopital Europeen Georges Pompidou;
- HMOD,
hypertension mediated organ damage;
- HT,
hypertension;
- LVH,
left ventricular hypertrophy;
- MDRD,
modification of diet in renal disease;
- MI,
myocardial infarction;
- PP,
pulse pressure;
- PWV,
pulse wave velocity;
- SBP,
systolic BP;
- TIA,
transient ischemic attack.
PATIENT CONSENT
Not required. However, patients should express that they are not opposed to participate to the protocol.
SUPPLEMENTARY MATERIAL
Supplementary data related to this article can be found at
Footnotes
REFERENCES
Cite this article
TY - JOUR AU - Stephane Laurent AU - Gilles Chatellier AU - Michel Azizi AU - David Calvet AU - Gabriel Choukroun AU - Nicolas Danchin AU - Pascal Delsart AU - Philippe Gosse AU - Gerard London AU - Jean-Jacques Mourad AU - Bruno Pannier AU - Helena Pereira AU - Dominique Stephan AU - Pierre Boutouyrie AU - On Behalf of SPARTE Investigators PY - 2020 DA - 2020/07/17 TI - Protocol of the SPARTE Study: A Strategy for Preventing Cardiovascular and Renal Events based on ARTErial Stiffness JO - Artery Research SP - 250 EP - 260 VL - 26 IS - 4 SN - 1876-4401 UR - https://doi.org/10.2991/artres.k.200711.001 DO - 10.2991/artres.k.200711.001 ID - Laurent2020 ER -