
Shear stress, inflammation and Atherosclerosis
- DOI
- 10.1016/j.artres.2010.03.002How to use a DOI?
- Keywords
- Shear stress; Inflammation; Atherosclerosis
- Abstract
Background: Atherosclerosis is the disease with one of the largest mortalities of the Western world. An important cause of this high mortality rate is rupture of atherosclerotic plaques and subsequent total blockage of the vessel by thrombus. Because, plaque rupture has been associated with a distinct plaque phenotype there is a growing need for understanding the pathogenesis of plaque composition. In this manuscript, we will discuss the new hypothesis that blood flow and lipid driven inflammation are intimately related to each other and that shear stress induced expression of chemokines and the resulting coordinated homing of inflammatory cells influence plaque composition in such a way that vulnerable plaque may be induced. As vulnerable plaques are intimately related to the high mortality of atherosclerosis, it re-emphasis the important role blood flow has in atherosclerosis.
- Copyright
- © 2010 Published by Elsevier B.V. on behalf of Association for Research into Arterial Structure and Physiology. All rights reserved.
- Open Access
- This is an open access article distributed under the CC BY-NC license.
Introduction
Atherosclerosis is the disease with the highest mortality in the Western World. Despite its large socio-economical impact, the underlying mechanisms are only partially known. While lipid-driven mechanism has been studied, over the last decade two concepts – lipid driven inflammation and blood flow/shear stress – have gained considerable interest as complementary explanations for plaque formation.
The hypothesis which states that there is a relation between local blood flow patterns and atherosclerosis is based upon the observation that plaques are not evenly distributed over the arterial system. Sites with a predilection for plaque development are at or near side branches where blood flow is non-uniform, or at the lesser curvature of bends where the velocity of blood particles is relatively low. The endothelial layer senses variations of blood flow through the effects of shear stress. Shear stress (τ N/m2 or Pascal (Pa)) arises from the friction between two virtual layers in a fluid, and is induced by the difference in movement of the two layers (dv/dr s−1; in the case of a cylindrical tube) and the “roughness” (or viscosity Pa.s) between these layers (τ = dv/dr*η).
Shear stress induces plaque formation and determines plaque composition
Shear stress has previously been correlated with plaque location in animals and humans before.1–4 The correlation between local shear stress and plaque location found by others1–3 and us,5 merely indicates an association between these factors. Associative relationships may be explained by a variety of factors. Indeed for the observations made between plaque location and shear stress at a single time point, it is unclear whether the disease induces the shear stress pattern or whether the shear stress pattern induces the disease. A stronger conclusion may be drawn when the shear stress pattern at a certain time point predicts the plaque distribution at a later time point. We have performed such a study in coronary arteries of patients after stent placement. In that study the shear pattern was predictive of neo-intima hyperplasia. However, the interpretation is still cumbersome, as multiple reasons may offer an explanation for this observation, including a redistribution of LDL particles towards the endothelium in low shear stress regions, different stretch patterns in low shear stress regions, and more stem cells adhesion in low shear stress regions. In order to provide evidence for a causal role of blood flow on plaque formation, we have developed an in vivo method to induce two well-accepted pro-atherogenic velocity profiles, i.e. a low, forward velocity/shear stress field and a low, oscillatory velocity/shear stress field. These two fields will be loosely described in this application as low and oscillatory shear stress fields or regions. When this method was applied to straight arterial carotid arteries of hypercholesteremic ApoE deficient mice, we measured an increased lipid accumulation both within the low shear stress region and the oscillatory shear stress region, albeit the low shear stress region exhibited a much larger effect (Fig. 1). Further analysis revealed that plaques in the low shear stress region were characterised by a large lipid core, a thin fibrous cap, a paucity of smooth muscle cells in the cap and by accumulation of inflammatory cells in the fibrous cap. This plaque composition in humans has been defined by Virmani et.al.5 as thin cap fibroatheroma (TCFA), which has been subsequently shown the phenotype responsible for plaque rupture. The presence of TCFAs in low shear stress regions in mouse carotid arteries is interesting as it identifies shear stress as key player in TCFA formation. The plaques in the oscillatory shear stress region displayed – in contrast – characteristics of fibrous plaques (Fig. 1).6,7

Description of the method to induce a vulnerable plaque. A home made device (lower left corner) is placed around the carotid artery of an ApoE −/− mouse placed on a high cholesterol diet. The carotid arteries remain free of plaque when flow through the vessel remains unchanged (upper left corner) but after 9 weeks of cast placement plaques develop upstream (low shear stress region) and downstream (oscillatory shear stress region) of the device. The compositions of the low shear stress induced and oscillatory shear stress induced plaques are different with a vulnerable phenotype upstream and a stable phenotype downstream of the device.
Low shear stress induced plaques display a characteristic chemokine pattern
One of the striking features of the low-shear-induced plaques is the high inflammatory state (unpublished findings and8). Low shear stress modulates the well-studied cascade model of homing of inflammatory cells,9–11 by increasing the expression of several adhesion and leukocyte-activating factors, resulting in increased capture of circulating leukocytes, reduction of rolling speed of inflammatory cells over the endothelium and increase in leukocyte arrest. Therefore, we tested whether this mechanism explains the different plaque composition in the two regions. Our first study revealed an increased expression level of several pro-inflammatory genes including VCAM-1, ICAM-1, and E-selectin in the low shear stress region,9,12–15 confirming the activation of the cascade model in that region. To further evaluate this model, we tested the expression of several chemokines reported to play a role in atherosclerosis.13 From this genomic screen, four chemokines were identified that were either expressed during stable and unstable plaque progression (MCP-1), or during low-shear-induced vulnerable plaque development (IP-10 and GRO-α), or in the established plaque with a vulnerable phenotype (Fractalkine; Fig. 2).
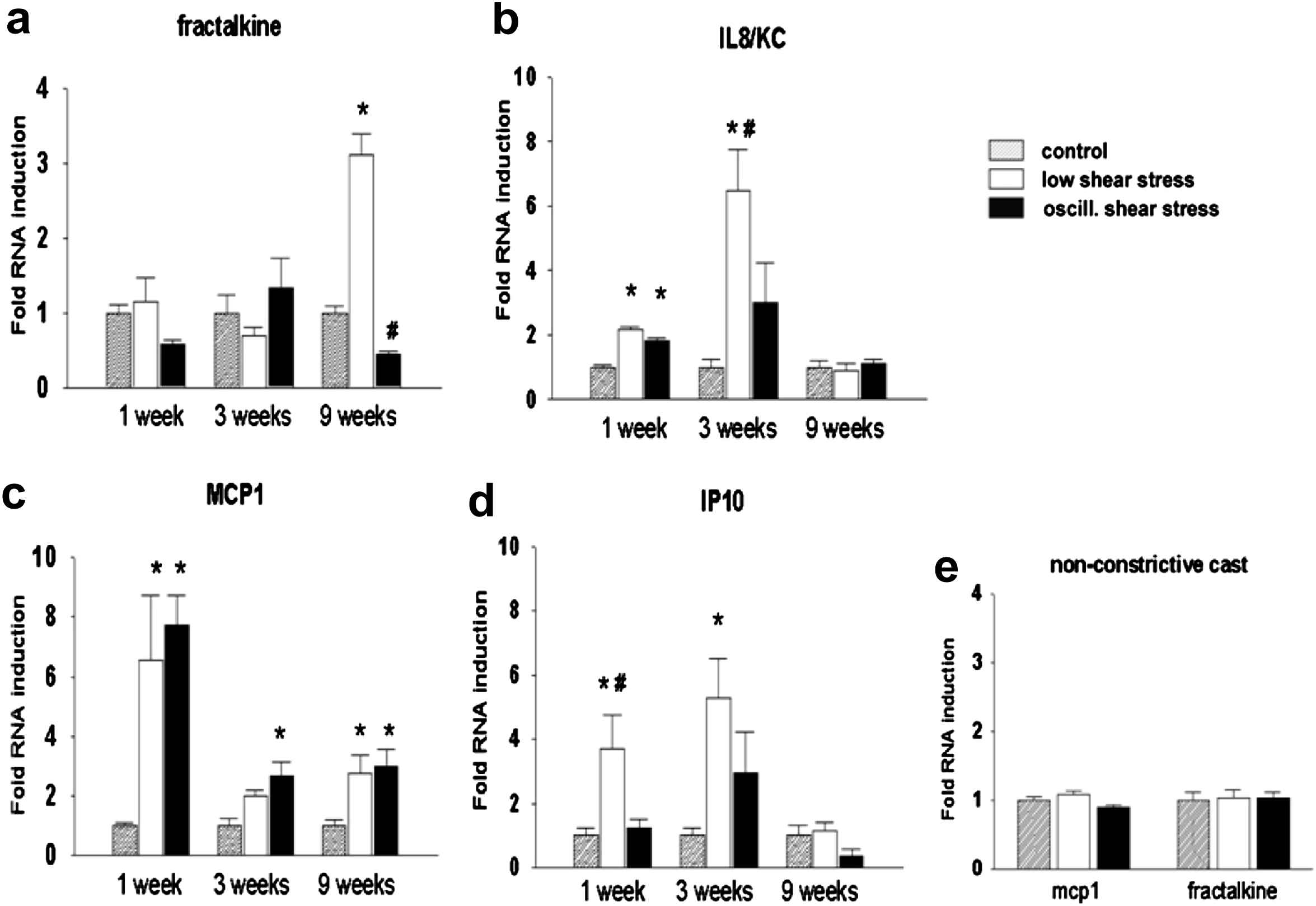
Expression pattern of Fractalkine (a), IL8 (b), MCP1 (c), IP10 (d) and control casts (e) in three different regions (Control, low shear stress and oscillatory shear stress regions) for three different time points (1 week, 3 weeks and 9 weeks). The three time points signify endothelial dysfunction (1 week), fatty streak formation (3 weeks) and complex plaques with a vulnerable phenotype (9 weeks). Note that Fractalkine, IP10 and GRO-α (IL8/KC) are differentially up regulated both in time and in location (see text for further details).
Based on these findings we inferred that a combination of at least these four chemokines is necessary to explain the low shear stress induced vulnerable plaque formation (Fig. 3). In essence, we propose that these four chemokines coordinate temporarily and spatially the uptake of a variety of inflammatory cells that become activated while being in close proximity of each other. We propose that only then, sufficient amount of cytokines are generated that will modify plaque composition leading to a vulnerable phenotype.

A proposed mechanism to explain vulnerable plaque formation. In the initial phase MCP1-CCR2 is activated attracting Ly6C + CCR2+CX3CR1- monocytes leading to the accumulation of classically activated macrophages and foam cells. This is followed by Th1 cell accumulation (through IP10, MIG and I-TAC) producing IFN-γ keeping activation of the macrophages high. Finally, Ly6C-CCR2-CX3CR1+ monocytes are attracted leading to the accumulation of dendritic cells (DCs) and increased activation of foam cells. This will lead to large amount of cytokine production, MMP production and tissue destruction. If local wall stress is sufficiently high these weak spots will lead to plaque rupture and myocardial infarction or stroke.
Initially, we studied Fractalkine because this chemokine was differentially expressed in the vulnerable plaques (Fig. 2). Furthermore, a recent study indicated that Fractalkine gene-targeted. mice exerted large effects in the Brachiocephalic Artery, an accepted location for plaques with a vulnerable morphology.13 We first confirmed that the differentially increased gene expression levels of Fractalkine were associated with concordant changes of protein levels, and with the presence of CX3CR1 receptors on macrophages. Subsequently, we showed that inhibition of Fractalkine reduced atherosclerotic plaque formation, reduced macrophage accumulation and increased collagen content, specifically in the vulnerable region. In summary, Fractalkine is specific for vulnerable plaques and modifies its plaque composition towards a stable phenotype.
One of the other chemokines, interferon-induced protein (IP-10; CXCL10) is a relative early differentially up regulated chemokine (Fig. 2) and is a chemo attractant for (activated) T-cells, monocytes and natural killer cells, resulting in migration of the cells into peripheral tissues. IP-10 is expressed by endothelial cells, smooth muscle cells and macrophages in plaques, identifying this chemokine as a functional candidate for (early) vulnerable plaque formation.16 We tested this hypothesis by measuring plaque morphology at 9 weeks of cast placement after inhibition of IP10 in the first three weeks of vulnerable plaque formation. Interestingly, IP10 inhibition increased collagen content, without affecting lipid and macrophage content of the plaques. Furthermore, IP10 inhibition reduced the activation level of macrophages in the vulnerable plaques, and increased the collagen content of the plaques. These findings are in accordance with the idea that IP10 orchestrates Th1 uptake and thereby modifies the activation level of macrophages without affecting the homing of this cell type (see Figs. 3 and 4).
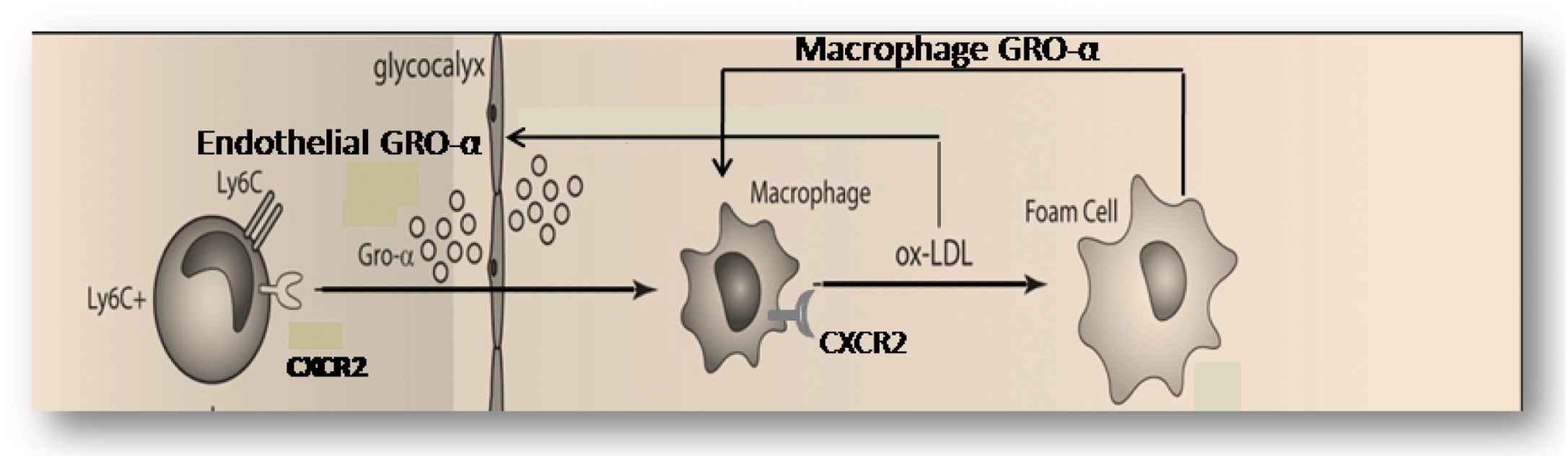
A detailed hypothesis obtained from the upper part of Fig. 3, explaining the two positive feedback loops for vulnerable plaque formation. In the first mechanism, ox-LDL induces macrophage-dependent GRO-α production and this GRO-α attract more CXCR2-positive macrophage to the site with ox-LDL accumulation. In the second mechanism, sub-endothelial ox-LDL and high shear stress both produce up-regulation of endothelial GRO-α increases local macrophage accumulation and (new) ox-LDL formation.
Low shear stress induced plaques display plaque haemorrhages and disruption after angiotensine-infusion
The definition of the composition of vulnerable plaque – as described above- has been obtained from post-mortem studies in patients.6,7 While these approaches certainly produce important information, it is unclear whether TCFAs are indeed more prone to rupture. Hence, we started up experiments to bring the TCFAs in the mouse to rupture by generating an increase of blood pressure by infusing Angiotensin II.17,18 As a marker for plaque rupture we defined plaque haemorrhages.19 Infusion of Angiotensin II over a period of three weeks immediately prior to the existence of TCFA, increased plaque haemorrhage from 30% to 80% only in the TCFA region and not in the stable region. In order to study the underlying mechanism we developed a MMP functional assay.17 Angiotensin II infusion increased collagenase activity in the TCFA region, predominantly by up-regulating MMP8 and MMP13 activity (Fig. 5), thereby increasing breakdown of the extracellular matrix inducing potential weak spots and increasing the susceptibility to rupture after increasing blood pressure.17 Whether this was solely due to the blood pressure augmentation induced by Angiotensin II or an effect on inflammation is currently unknown17
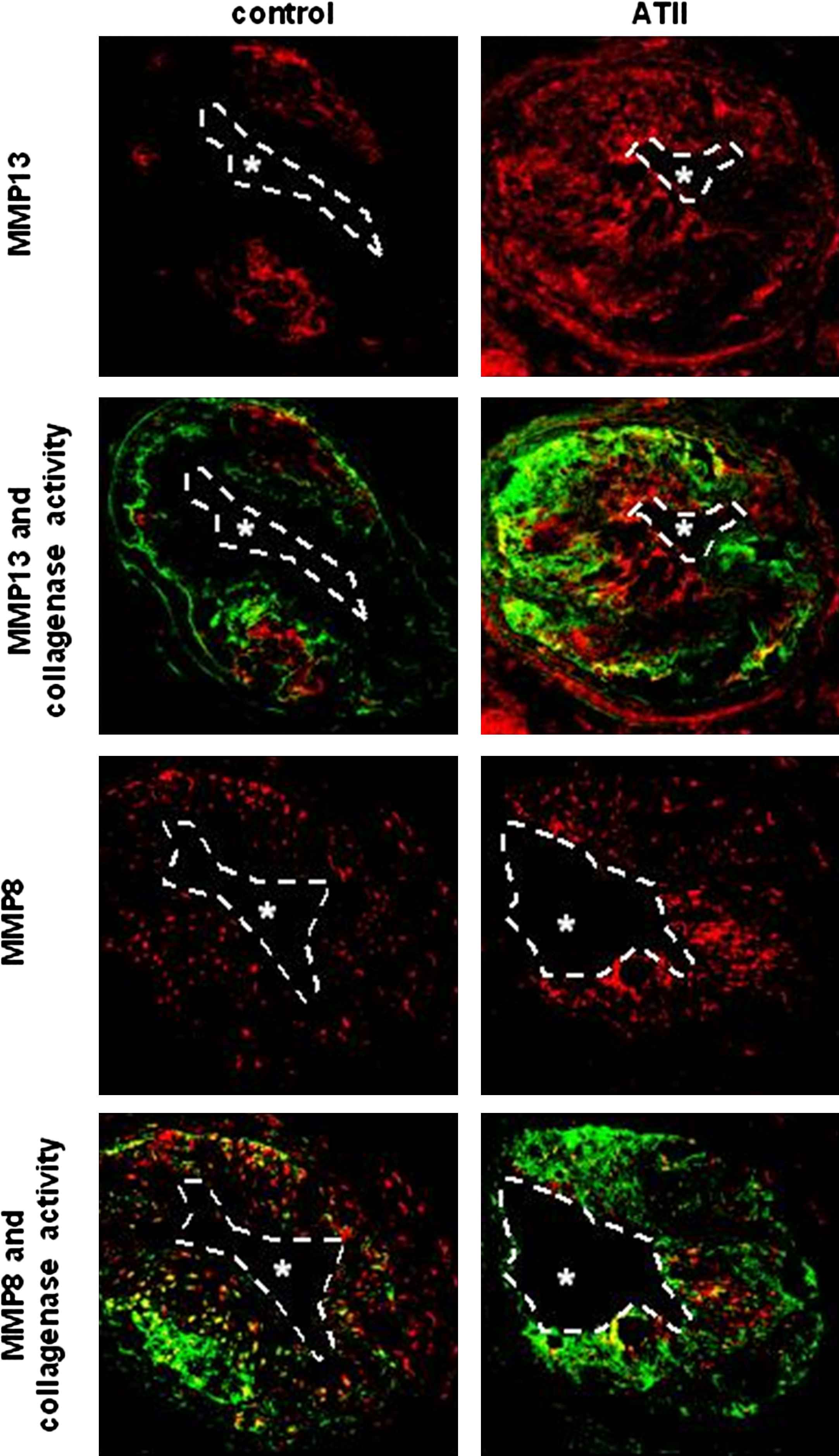
Co-localisation between MMP8, MMP13 and collagenase activity in low shear stress induced plaques treated with and without Angiotensin II infusion (ATII). Note the increased collagenase activity co-localised with the increased MMP8 and with increased MMP13 levels.
Low shear stress induces longitudinal plaques heterogeneity similar to that of humans
Longitudinal heterogeneity of plaques has hardly been studied, but the few studies that have been published indicate a predominant accumulation of inflammatory cells upstream of the plaques.4,17,20,21 In order to study whether the low shear stress induced plaque comply with findings published in patients, we studied longitudinal heterogeneity in the current animal model. Indeed, upstream accumulation of activated macrophages was more apparent upstream than downstream in our animal models,4,17,20,21 which is in accordance with observations in post-mortem studies and in studies with virtual histology in patients.22 The underlying mechanisms are currently unknown but the observations indicate that a spatial organisation of inflammation plays an important role in addition to the temporal organisation of inflammation.
In conclusion, our studies indicate that in addition to the current risk factors blood flow may initiate and sustain atherosclerotic plaque formation, probably by modifying the inflammatory response to accumulated lipids. Blood flow not only plays a role in atherogenesis, but also in plaque composition. It appears that low shear stress induces vulnerable plaques, while oscillating shear stress induces stable plaques when high cholesterol levels are present.
Acknowledgement
The funding of the
References
Cite this article
TY - JOUR AU - Rob Krams AU - Simon Cuhlmann AU - Nicolas Foin AU - Paul Evans PY - 2010 DA - 2010/04/28 TI - Shear stress, inflammation and Atherosclerosis JO - Artery Research SP - 41 EP - 46 VL - 4 IS - 2 SN - 1876-4401 UR - https://doi.org/10.1016/j.artres.2010.03.002 DO - 10.1016/j.artres.2010.03.002 ID - Krams2010 ER -