
Arterial structure and function in end-stage renal disease
- DOI
- 10.1016/j.artres.2007.06.001How to use a DOI?
- Keywords
- Cardiovascular disease; End-stage renal disease; Arterial function; Arterial structure
- Abstract
Cardiovascular disease (CVD) is a major cause of morbidity and mortality in patients with end-stage renal disease (ESRD). The risk of CVD disease in these patients appears to be far greater than in the general population and even after the stratification for age, gender, race and the presence or absence of diabetes, cardiovascular mortality in dialysis patients is about 10 to 20 times higher than in the general population. As a diagnostic categorie, CVD includes two principal conditions, i.e., cardiac complications stricto sensu, and vascular complications. These two complications are interrelated and arterial alterations can be the primary reason for the development of cardiac complications. Macrovascular disease develops rapidly in ESRD patients and is responsible for the high incidence of congestive heart failure, left ventricular hypertrophy (LVH), ischemic heart disease, sudden death, cerebrovascular accidents and peripheral artery diseases. Although the most frequent cause of these complications is occlusive lesions due to atheromatous plaques, many complications arise in ESRD patients in the absence of clinically significant atherosclerotic disease. Atherosclerosis, disease characterized by the presence of plaques, represents only one form of structural response to metabolic and hemodynamic alterations which interfere with the “natural” process of aging. The spectrum of arterial alterations in ESRD is broader, including large artery remodeling and changes in viscoelastic properties of arterial walls. The consequences of these alterations are different from those attibuted to plaques. While atheromatous lesions alter principally the conduit function of arteries and perfusion of tissue and organs downstream the lesions, non-atheromatous remodelling result principally in changes in dampening function of arteries, characterized by stiffening of arterial walls and with deleterious effects on the left ventricle and coronary perfusion. Arterial stiffening in ESRD patients is multifactorial at origin with extensive arterial calcifications as an important covariate.
- Copyright
- © 2007 Association for Research into Arterial Structure and Physiology. Published by Elsevier B.V. All rights reserved.
- Open Access
- This is an open access article distributed under the CC BY-NC license.
Introduction
Cardiovascular disease is a major cause of morbidity and mortality in patients with end-stage renal disease (ESRD).1 Epidemiological and clinical studies have shown that damage of large conduit arteries is a major contributing factor responsible for the high incidence of congestive heart failure, left ventricular hypertrophy (LVH), ischemic heart disease, and peripheral artery diseases. Although atherosclerosis, characterized by the presence of plaques and occlusive lesions, is the most frequent underlying cause of these complications, the spectrum of arterial alterations in ESRD is broader, including large artery non-atheromatous remodeling and stiffening whose consequences are different from those attributed to the presence of atherosclerotic plaques.2
Arterial remodeling and arterial function: basic concepts
Arterial remodeling
An arterial wall is a complex tissue capable of structural and functional changes in response to atherogenic factors (atherosclerosis) or changes in hemodynamic.3 While acute changes in tensile or shear stress induce transient adjustments in vasomotor tone and in arterial diameter,4 chronic alterations of mechanical forces lead to changes in the geometry and composition of the vessel walls.5–8 The mechanical signals for arterial remodeling associated with hemodynamic alterations are the cyclic tensile stress and/or shear stress.5,9,10 Blood pressure is the principal determinant of arterial wall stretch and tensile stress. According to Laplace’s law, tensile stress (σ) is directly proportional to arterial transmural pressure (P) and radius (r), and inversely proportional to arterial wall thickness (h) according to the formula: σ = Pr/h. In response to increased blood pressure or arterial radius, tensile stress is maintained within the physiological range by thickening of the vessel wall.
Blood-flow alterations result in changes in shear stress - the dragging frictional force created by blood flow.6,11 Shear stress is the product of shear rate (i.e. the change in blood parabolic velocity profile per unit distance across the vessel radius), times blood viscosity. Thus, shear stress (τ) is directly proportional to blood flow (Q) and blood viscosity (η) and inversely proportional to the radius (r) of the vessel, according to the formula: τ = Qη/πr3. Changes of shear and tensile stresses are interrelated because any modification of arterial radius caused by alterations in blood flow and shear stress induces changes in tensile stress (unless the pressure varies in the opposite direction).
The process of transforming mechanical forces into remodeling of vascular system implies that there are “sensors” that detect and transmit physical forces to effector cells. Endothelial cells are strategically situated at the blood-vessel wall interface and are the principal candidates for the role of “sensors”.12,13 This mechanosensor activation results in the transduction of physical stimuli into a biochemical signal affecting arterial function through the generation of vasoactive substances,14–17 releasing and/or activating growth factors and adhesion molecules18,19 and extracellular matrix regulators.20–23 The characteristics of arterial remodeling depend largely on the nature of hemodynamic stimuli applied. Experimental and clinical data indicate that acute and chronic augmentations of arterial blood flow induce proportional increases in the vessel lumen, whereas decreasing flow reduces arterial inner diameter.6,24 Increased arterial inner diameter is usually accompanied by arterial wall hypertrophy and increased intima-media cross-sectional area (following increases in the radius and wall tension). Although the alterations of tensile and shear stresses are interrelated, changes of tensile stress primarily induce alterations and hypertrophy of the arterial media.3,25 Tensile stress and shear stress also influence the natural history of atherosclerotic lesions.6,26,27 Evidence that enhanced tensile stress is relevant to the pathogenesis of atherosclerosis comes from the observations that atherosclerotic plaques are virtually confined to systemic arteries where tensile stress is high, while the role of shear stress is demonstrated by the predilection of atherosclerosis for sites characterized by low average shear stress or turbulent flows.27–29 Atherosclerosis is uncommon in sites with high shear stress and in these locations, the endothelial cells are aligned in the direction of flow,30 thereby decreasing the effective resistance to friction.29,30 The physiological shear stress promotes the secretion and synthesis of nitric oxide, prostacyclin, antithrombotic and antigrowth factors that mediate atheroprotection, and survival of endothelial cells.14,16,30–33 At sites of low shear stress or turbulent flows (characterized by flow reversal and time-averaged shear stress approaching zero), endothelial cells secrete prothrombotic and progrowth factors, and trigger endothelial apoptosis.29,30–33
Arterial functions
The arterial system has two distinct, interrelated hemodynamic functions: (1) to deliver an adequate supply of blood from the heart to peripheral tissues, as dictated by metabolic activity (conduit function); and (2) to dampen blood pressure oscillations caused by intermittent ventricular ejection (dampening function). Disorders of conduit function result from the narrowing of the arterial lumen with ischemia affecting the tissues and organs downstream, while disorders of the dampening function reflect alterations of arterial wall viscoelastic properties and have deleterious effects upstream on the heart and the arteries themselves.11,34
Conduit function of arteries
Conduit-function efficiency is the consequence of the very low resistance of large arteries to flow and is primarily dependent on the diameter of the arterial lumen. The conduit function is highly efficient and can accommodate increases in flow to some tissues, like muscle, by perhaps 10-fold. This physiological adaptability is mediated through acute changes of arterial flow velocity and/or diameter changes which are dependent on the endothelium response to alterations in shear stress.6,10 Under conditions of long-term flow overload, the arterial diameters are enlarged and baseline arterial conductance is increased.4,6–8 The principal long-term alterations of conduit function occur through narrowing or occlusion of arteries with restriction of blood flow and resulting ischemia or infarction of downstream tissues. Atherosclerosis, characterized by the presence of plaques and arterial narrowing, is the most common occlusive vascular disease that disturbs conduit function. Owing to the large luminal area of conduit arteries, basal blood flow remains unchanged until the lumen diameter is narrowed by 50–60%. Beyond 70–80% reduction of the lumen diameter (critical stenosis), basal blood flow is reduced as is the ability to increase flow during activity.11,34
Dampening function of arteries
The role of arteries is to dampen the pressure oscillations resulting from intermittent ventricular ejection (“windkessel” effect) and to transform the pulsatile flow of arteries into the steady flow required in peripheral tissues and organs.11,34 The large arteries can instantaneously accommodate 50–60% of stroke volume (40% is forwarded directly to peripheral tissues) distending the walls. Approximately 10% of the energy produced by the heart is diverted for the distension of arteries and “stored” in the walls. During diastole, most of the stored energy recoils the aorta, squeezing the stored blood forward into the peripheral tissues, thereby ensuring continuous perfusion of organs and tissues. For the dampening function to be efficient, it is essential that the energy necessary for arterial distension and recoil be as low as possible, i.e., for a given stroke volume, the pulse pressure (ΔP) should be as low as possible. The efficiency of the Windkessel function depends on the viscoelastic properties of arterial walls and the geometry of the arteries, including their diameter and length.11,34 The ability of arteries to accommodate the volume ejected by the LV instantaneously can be best described in terms of stiffness of the aorta or an individual artery. This term express the given transmural pressure as a function of contained volume of the vasculature (total or segmental) over the physiological range of pressure. In physiology, stiffness or elastance (E) is defined as the change in pressure (ΔP) due to a change in volume (ΔV), that is E = ΔP/ΔV. Stiffness/elastance represents the instantaneous slope of the pressure-volume relationship. The reciprocal value of E is compliance (C = ΔV/ΔP). Because it is composed of a “mixture” of smooth muscle cells and connective tissue, containing elastin and collagen fibers, the pressure volume relationship is nonlinear. At a low distending pressure, the tension is borne by elastin fibers, whereas at a high distending pressure, the tension is predominantly borne by less extensible collagen fibers and the arterial wall becomes stiffer (less compliant).11,34 To facilitate comparisons of viscoelastic properties of structures with different initial dimensions, viscoelastic properties of arteries could be expressed as distensibility (Di), i.e. pressure/volume relationship relative to the initial/baseline volume as Di = ΔV/ΔPV, where ΔV/ΔP is compliance and V is the initial volume. In contrast to distensibility or compliance which provides information about the “elasticity” of the artery as a hollow structure, the elastic incremental modulus (Einc; Young’s modulus) provides direct information on the intrinsic elastic properties of the materials that compose the arterial wall independent of vessel geometry. Owing to the inhomogeneity of the viscoelastic properties of successive arterial segments and the effect of arterial wave reflections, pulse and systolic pressures are amplified from the aorta to the peripheral arteries.35–38 To determine the elastic properties of arterial segments accurately, the “local” pulse pressure must be measured and taken into consideration. Arterial stiffness (distensibility) can be evaluated by measuring the pulse wave velocity (PWV) over a given arterial segment.11,39,40 PWV increases with arterial stiffness.39,40 The dampening function is altered by decreased distensibility and stiffening of arterial walls. The principal consequences of arterial stiffening are an increase in systolic and decrease in diastolic pressures with resulting high pulse pressure.34,41–43 Pulse pressure depends on the interaction between LV ejection (stroke volume and duration of systole) and the physical properties of the arterial system that influence pulse pressure by two mechanisms. The first, direct mechanism involves the generation of a higher pressure wave (incident or forward traveling wave) by the LV ejecting into a stiff arterial system, and decreased diastolic recoil resulting in lowered diastolic pressure. This forward-traveling (incident) pressure wave is reflected at any point of structural and functional discontinuity in the arterial tree, thereby generating a reflected wave traveling backwards towards the ascending aorta.38,41,42,44 Incident and reflected pressure waves interact constantly and their sum gives the measured pulse pressure wave. The final amplitude and shape of the measured pulse pressure wave are determined by the phase relationship (the timing) among the component waves.38,44 Peripheral arteries are close to reflection sites, and the incident and reflected waves in these arteries are in phase and, thus, produce an additive effect. The ascending aorta and central arteries are distant from reflecting sites and, depending on the PWV and arterial length, the return of the reflected wave is variably delayed and thus the incident and reflected waves are not in phase.11,34,38,44 The timing of incident and reflected pressure also depends on the duration of LV ejection (heart rate) and transit time of pressure waves to and back from reflecting sites.11,34,44,45 In subjects with distensible arteries and low PWV, the reflected waves affect central arteries during diastole after LV ejection has ceased. This timing is desirable, since the reflected wave causes an increase in ascending aortic pressure during early diastole resulting in aortic systolic and pulse pressures which are lower than in peripheral arteries (only mean blood pressure is almost constant throughout the arterial system).35–44 This situation is physiologically advantageous since the increase in early diastolic pressure has a boosting effect on coronary perfusion without increasing LV afterload. The desirable timing is disrupted by increased PWV due to arterial stiffening. With increased PWV, the reflecting sites appear “closer” to the ascending aorta and the reflected waves occur earlier, being more closely in phase with incident waves in this region. The earlier return means that the reflected wave affect the central arteries during systole rather than diastole, thus amplifying aortic and LV pressures during systole and reducing aortic pressure during diastole. By favoring early wave reflections, arterial stiffening increases peak- and end-systolic pressures in the ascending aorta increasing myocardial pressure load (left ventricular hypertrophy) and oxygen consumption, decreasing the diastolic blood pressure and subendocardial blood-flow.46–48
The dampening function of the arterial tree is altered primarily during the aging process and in conditions associated with “sclerotic” remodeling of arterial walls, i.e., associated with increased collagen content and modifications of extracellular matrix (arteriosclerosis).40,47–49 Arteriosclerosis is primarily manifest as medial degeneration that is generalized throughout the thoracic aorta and central arteries, causing dilatation, diffuse hypertrophy, and stiffening of the arteries.34,40,41,47,50 Age-related arterial alterations leading to stiffening are heterogeneous, being more pronounced in the aorta and central, elastic-type, capacitative arteries than in the peripheral muscular-type limb arteries.40
Arterial remodeling and function in ESRD patients
Atherosclerosis
Atherosclerosis and arterial occlusive lesions with alterations of conduit function occupy an important place in the mortality of ESRD patients.1 The high incidence of atherosclerosis-related complications led Lindner and colleagues51 to hypothesize that atherogenesis is accelerated in chronic hemodialysis patients. While ESRD produces pathological factors essentially unique to uremia, including calcium-phosphate alterations, malnutrition and activation of cytokines, these factors are additive to the number of traditional risk factors such as age, dyslipidemia(s), hypertension, smoking, diabetes, male gender, and insulin resistance. Risk factors present before ESRD might be of primary importance52,53 and ESRD patients have significant vascular lesions before initiating dialysis. In many patients the generalized atherosclerosis can be the primary cause of renal failure (ischemic renal disease, cholesterol embolization, etc.).54 It remains a matter of debate whether or not the atherogenesis is accelerated in ESRD patients but the nature of atherosclerotic plaques is different in hemodialysis patients and the general population. Indeed, ultrasonographic studies have shown a much higher prevalence of calcified plaques in ESRD patients than in age-matched controls in whom soft plaques are more frequent.55 Hemodynamic alterations could also favor the development of atherosclerotic occlusive lesions. Hypertension is a frequent complication in chronic renal diseases, and an association between high blood pressure and occlusive lesions was found in chronic hemodialysis patients.56 The role of shear stress in the development of atherosclerosis in ESRD patients has not been specifically investigated, but shear stress is lower in ESRD patients associated with endothelial dysfunction and arterial remodeling.57–59
Arteriosclerosis
The arterial system of ESRD patients undergoes remodeling that is characterized by dilatation and, to a lesser degree, arterial intima-media hypertrophy.2,58–61 In ESRD patients, this remodeling is associated with arterial stiffening2,58–62 due to alterations of the intrinsic properties of arterial wall materials (Einc) including those arteries free of atherosclerosis, such as upper arm arteries.60–63 The effect of increased Einc on the arterial stiffness is in part attenuated by the arterial dilatation, and the compliance of capacitive arteries is less altered than Einc.
The aging process is accompanied by progressive increase in the arterial diameters which compensate for progressive age-related stiffening of arterial walls. This age-related process is accelerated in ESRD patients and arteries are enlarged in ESRD patients in comparison with age-, sex-, and pressure-matched control subjects (Fig. 1).2,58,60 Arterial enlargement and increased stiffness are already observed in early stages of chronic kidney disease and at the onset of dialysis, suggesting that arterial remodeling takes place early in the course of renal failure.64 Outward remodeling with increased inner diameter is a vessel-wall adaptation to chronic blood flow change and is regulated by shear stress-induced changes of the endothelium. In ESRD these changes are typically observed in the brachial artery at the arteriovenous fistula site.65 Acutely, fistula creation increases blood flow, mean SS and BA diameter. With time, blood flow remains high and the diameter increases further but this modification did not result in SS restoration.65 In ESRD patients, the BA outward remodeling was also observed in BA not wearing A-V fistula.58,64 This outward remodeling was not associated with higher blood flow, but with an acceleration of age-associated increase in internal dimensions (Fig. 1), resulting in significant reduction in shear stress, due to low shear rate and anemia-associated low whole blood viscosity.58 (Fig. 2). As already mentioned laminar physiological fluid SS promotes endothelial cells survival and quiescence.31–33 Maintenance of a physiological laminar SS is crucial for normal vascular structure and function and exerts atheroprotective properties. Reduced shear stress in ESRD patients is associated with high circulating levels of endothelial microparticles,59 increased arterial stiffness (Fig. 3), and reduced endothelial-mediated flow-mediated dilation.58 Increased shear stress associated with improvement of anemia/WBV was associated with reduced arterial stiffening, improved flow-mediated vasodilation, and decrease in endothelial microparticles release.58,59
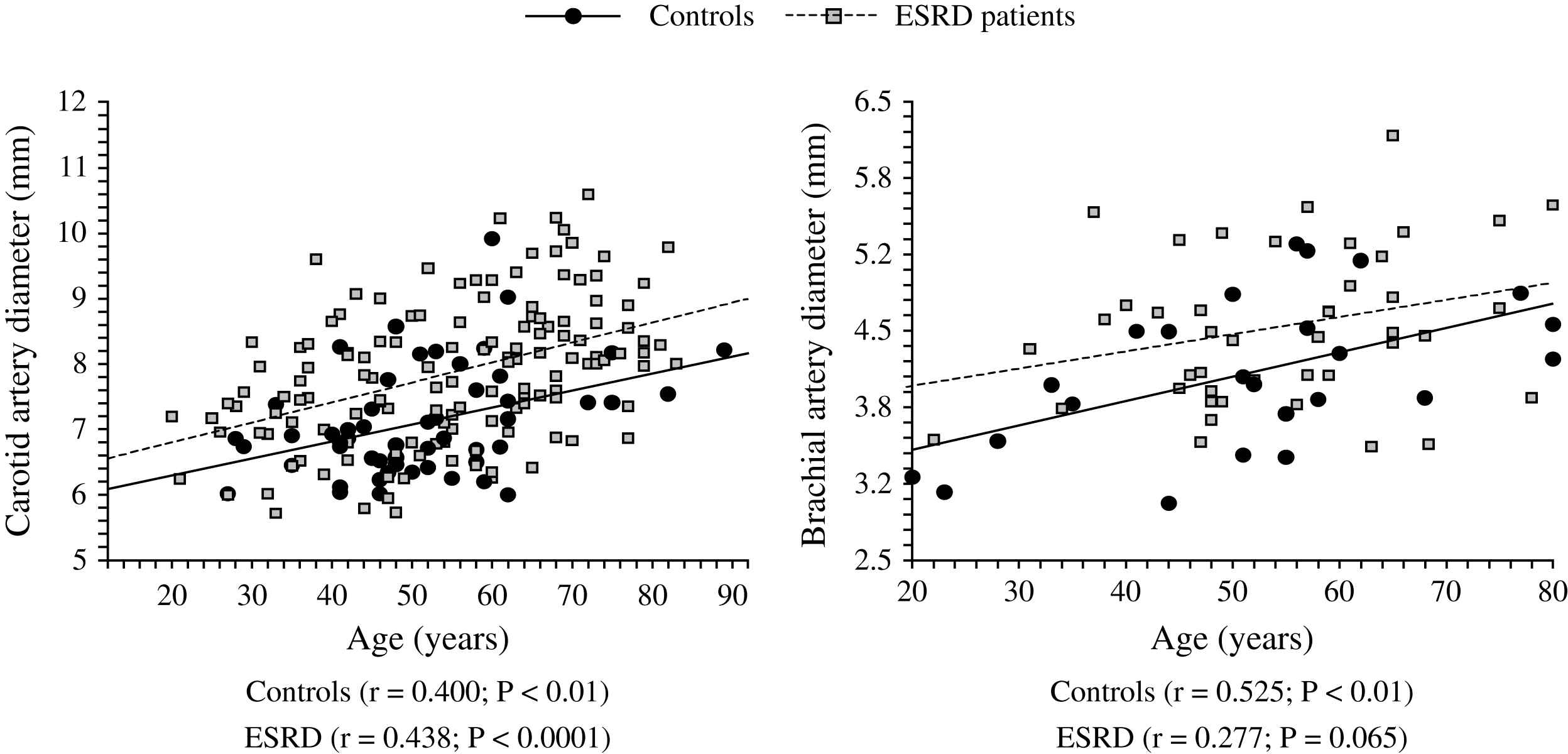
Correlations between common artery and brachial artery diameters and age in control population (●) and end-stage renal disease patients (□).
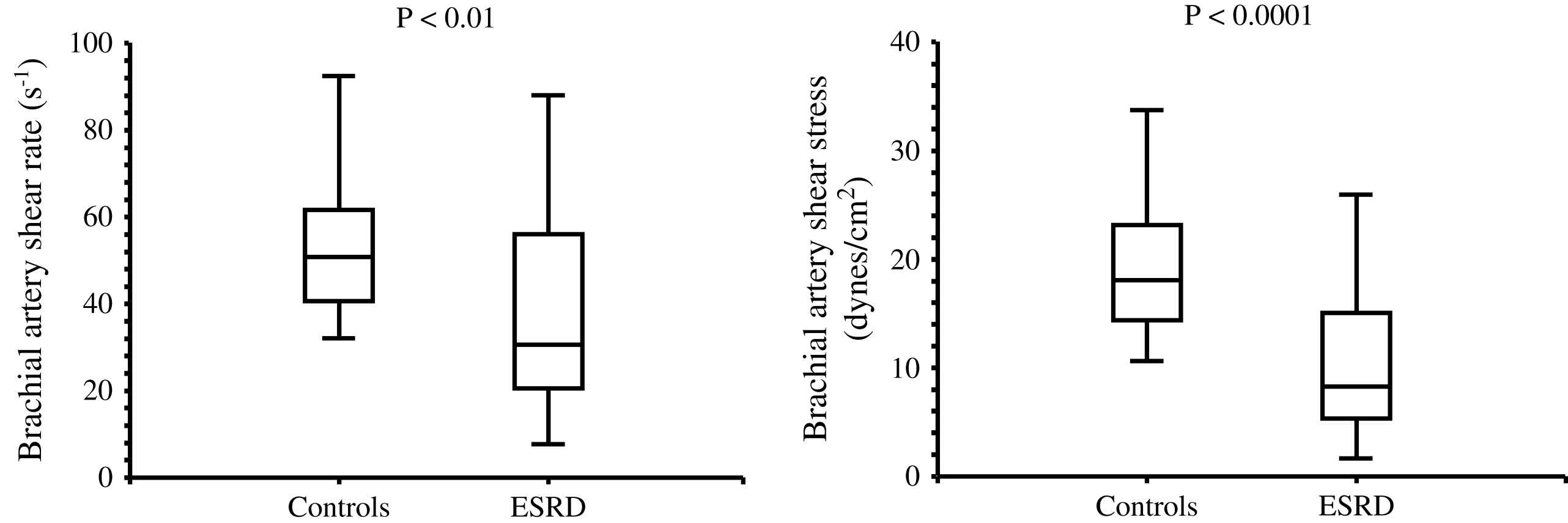
Brachial artery shear rate and shear stress in control population and end-stage renal disease patients.
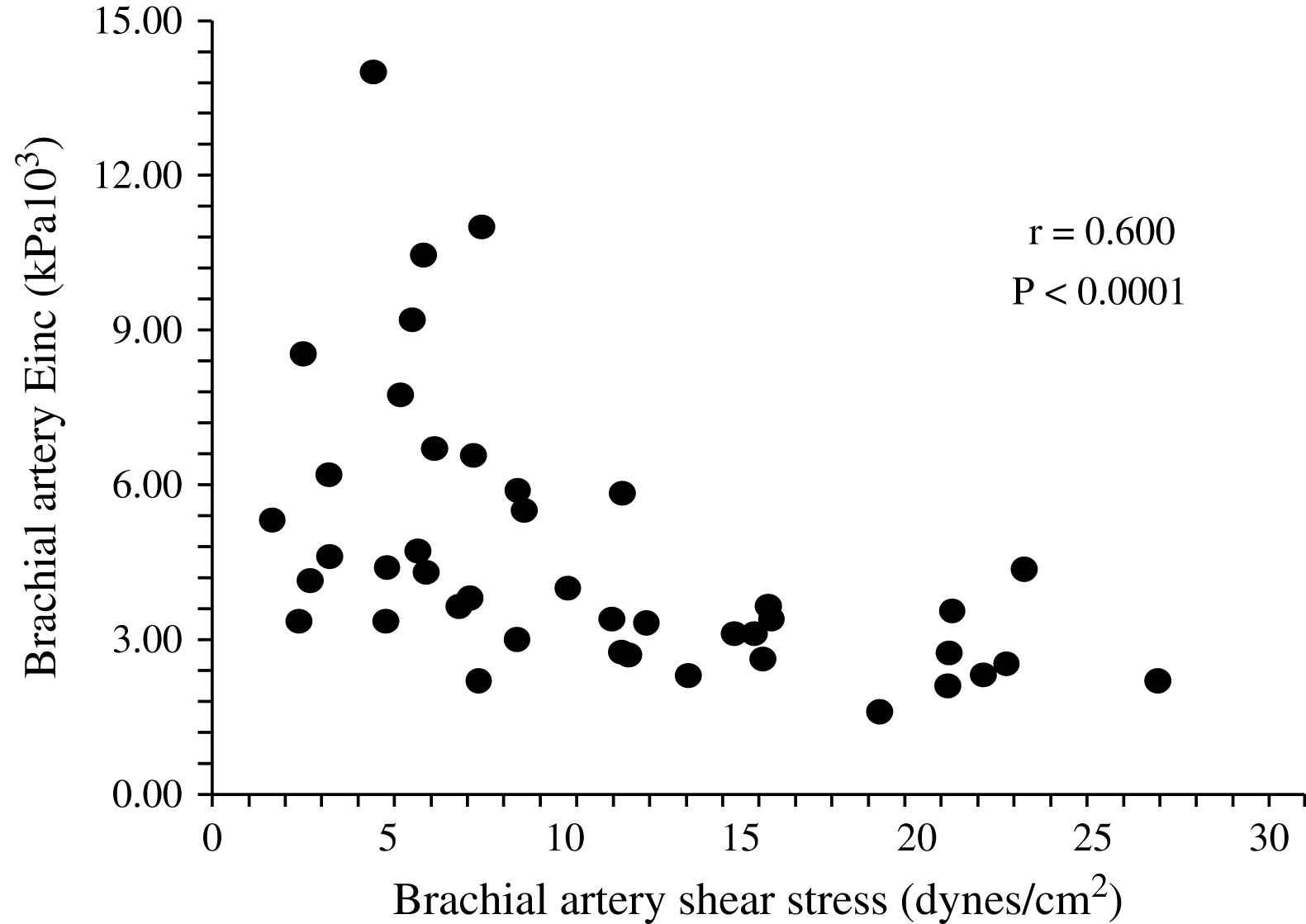
Correlation in end-stage renal disease patients between brachial artery shear stress and brachial artery incremental elastic modulus.
Unlike in blood pressure- and age-matched non-uremic patients, the arterial intima media thickness (or arterial wall cross-sectional area) is moderately increased in ESRD patients.2,60,66 By contrast, the intima-media thickness is proportional to changes in diameter, with wall-to-lumen ratio not different from that of non-uremic controls.2,58,64 According to Laplace’s law, arterial wall hypertrophy could be considered a response to increased circumferential tensile stress whereby wall tension is directly proportional to arterial radius. However, according to the same law, when the blood pressure increases, and regardless of the internal radius, the wall-to-lumen ratio should increase in order to normalize the tensile stress.11 This increase is observed in non-uremic populations but not in ESRD patients whose wall-to-lumen ratio in large conduit arteries is not related to pressure changes (Fig. 4). The difference between the observed relationships suggests that conduit arteries could have limited capacity to hypertrophy in response to pressure load. Arterial stiffness and distensibility is “pressure-dependent”11,34 and, in essential hypertensive patients, the decreased arterial stiffening is associated with higher distending blood pressure. In patients with essential hypertension, the arterial stiffening seems more associated with arterial wall thickening rather than modifications in intrinsic biomaterial stiffness. When adjusted for differences in blood pressure (i.e., under isobaric conditions), the arterial distensibility and/or elastic modulus of essential hypertensive subjects are more distensible (in muscular conduit arteries) or similar (in elastic capacitive arteries) to those observed in normotensive control subjects.67,68 These observations suggest that, in essential hypertension, the biomaterials are similar to that of normotensive subjects, with hypertrophy of the walls being due only to increased amount of wall biomaterials. This concept is different from the observation made in ESRD patients, in whom aortic and carotid artery stiffness is increased in comparison to age- and blood pressure-matched non-uremic subjects (Figs. 5 and 6).58,63 In ESRD patients, arterial hypertrophy is accompanied by alterations in the intrinsic elastic properties of arterial walls (increased Einc) that contribute to creating and amplifying the pressure load. This modification affects elastic and muscular-type arteries, including arteries free of atherosclerosis like the radial artery.63 The observation that the incremental modulus of elasticity is increased in ESRD patients more strongly favors altered intrinsic elastic properties or major architectural abnormalities like those seen in experimental uremia and the arteries of uremic patients, namely fibroelastic intimal thickening, calcification of elastic lamellae, increased calcium, increased extracellular matrix, and more collagen with relatively less of the elastic fiber content.69–71 Arterial wall properties are influenced by non specific factors, like age and body height, but also by factor(s) associated with the presence of uremia per se. The most frequently observed factors associated with arterial remodeling and functional alterations in ESRD seem to be alterations in calcium and phosphate metabolism.66,72–75 In hemodialyzed patients, arterial wall thickness and stiffness were found to be associated with mediacalcinosis (Fig. 7).74–76 No consistent and constant associations could be established between arterial remodeling and common atherogenic factors and association of arterial alterations with lipid abnormalities is not obvious and was found only irregularly. Some authors61,72 described a positive relationship between carotid intima media thickness and IDL or LDL cholesterol, these results were not constantly found in ESRD patients.
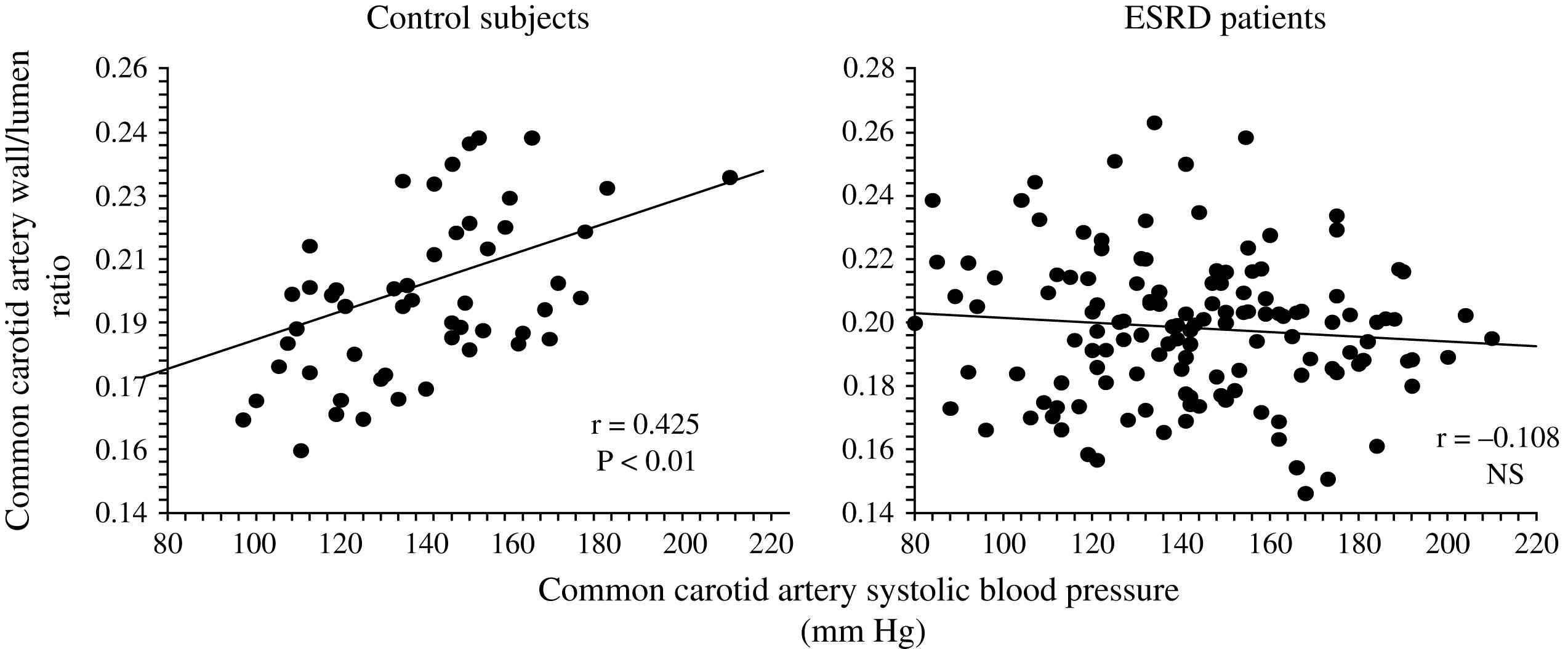
Correlations between common carotid artery wall-to-lumen ratio and carotid artery systolic blood pressure in control population and end-stage renal disease patients.

Correlations between common artery and brachial artery incremental elastic modulus (Einc) and age in control population (●) and end-stage renal disease patients (□).
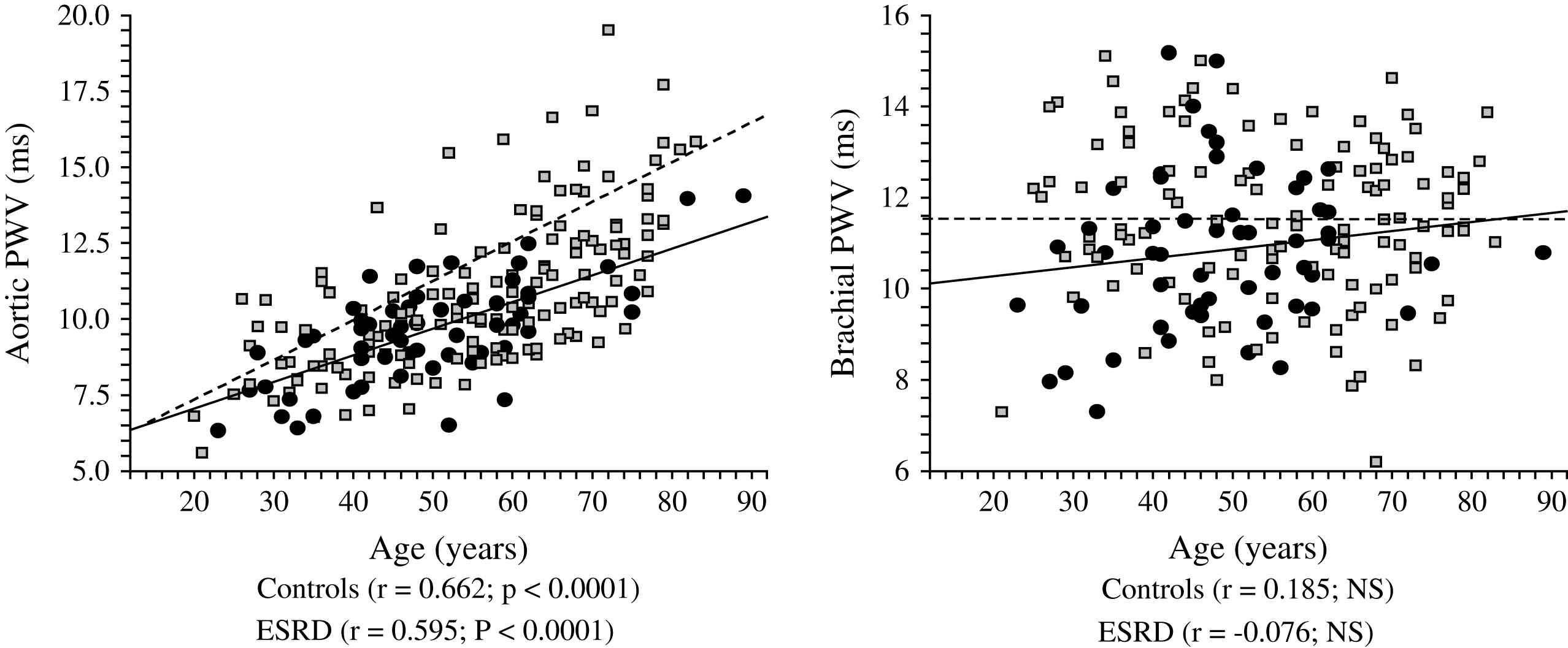
Correlations between aortic and brachial pulse wave velocities (PWV) and age in control population (●) and end-stage renal disease patients (□).
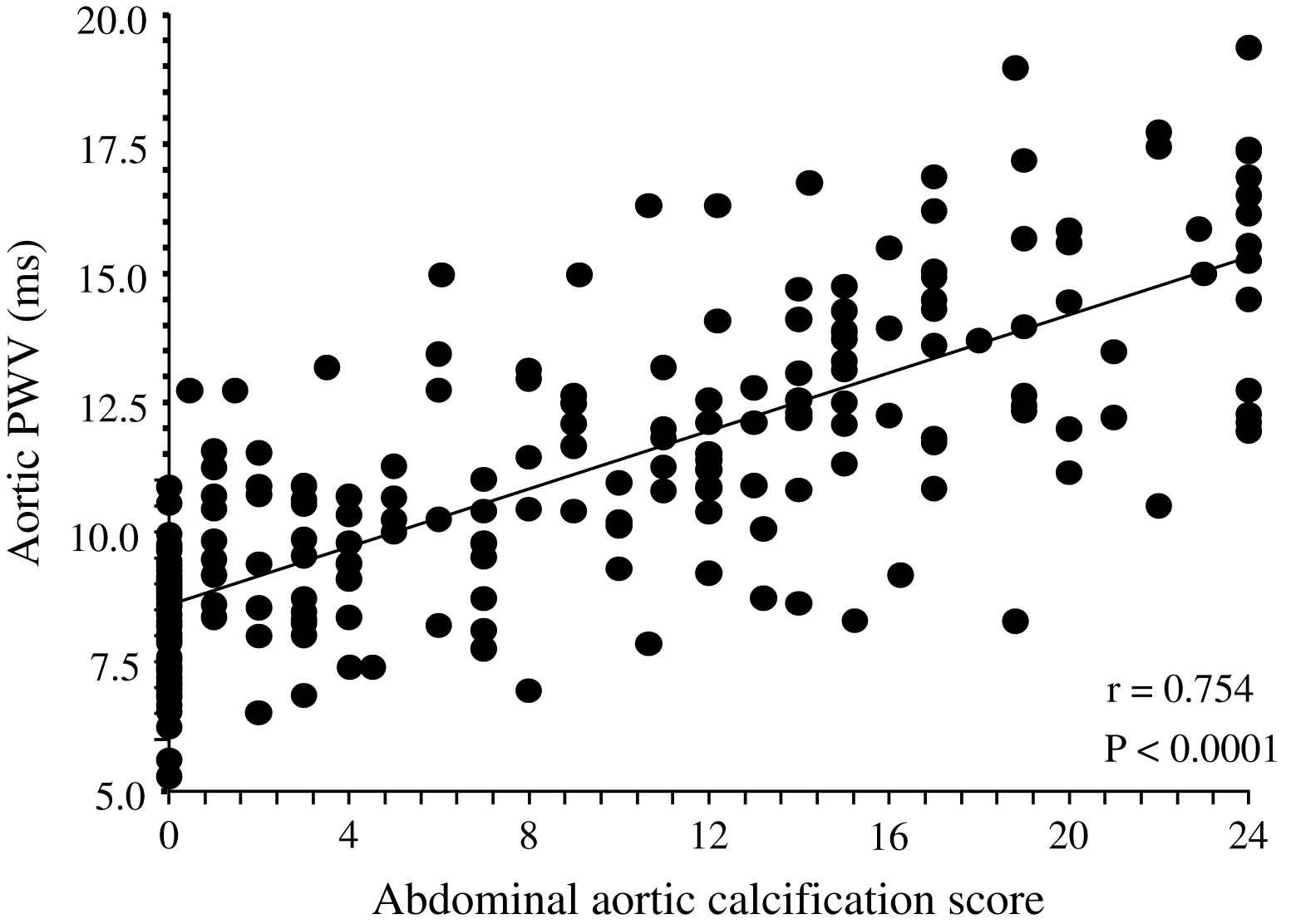
Correlation between abdominal aorta calcification score87 and aortic pulse wave velocity in end-stage renal disease patients.
The principal pathophysiological consequence of vascular alterations in ESRD is increased aortic stiffening with increased aortic PWV and early wave reflections responsible for abnormal augmentation in aortic and left ventricular pressure load.2,45,62 Arterial stiffening concerns principally aorta and large central arteries such as common carotid artery, while peripheral conduit arteries are less affected.2,61 The principal clinical consequences of these effects are increased systolic and pulse pressures, LV hypertrophy, and altered coronary perfusion. Previous studies showed that LV hypertrophy in ESRD is correlated with increased arterial stiffness and the intensity of wave reflections.2,45,77 The alterations in coronary perfusion in ESRD patients was suggested by the association of the large artery structure and function with decreased subendocardial viability index an index of the propensity for subendocardial ischemia in the absence of occlusive arterial lesions.2,45 Recent studies demonstrated that arterial stiffening and increased wave reflections are per se independent predictors of all-cause and cardiovascular death in ESRD patients as well as in the general population.78–81 PWV is a complex parameter integrating arterial geometry and intrinsic elastic properties. Based on Cox analyses, Blacher and colleagues showed that the principal factor associated with PWV as a predictor of cardiovascular and all-cause mortality in ESRD was increased elastic modulus.82 While the prognostic value of aortic PWV was demonstrated by several studies, in a recent study Pannier et al.83 have shown that to the difference with aortic PWV, peripheral arteries PWV(s) have no prognostic value (Fig. 8). Clinical studies involving essential hypertension and ESRD patients showed that antihypertensive drugs decrease aortic PWV to a large extent in response to BP lowering.84,85 This BP dependency of arterial stiffness offers the possibility to investigate whether the regression of arterial stiffness has a favorable effect on patient outcome. This possibility was studied in a follow-up study by Guérin et al.86 Their results showed that, in patients in whom PWV decreased in parallel with BP, survival was much better than in those ESRD patients with whose PWV was insensitive to decreased BP. This effect of PWV was totally independent of BP changes as such.
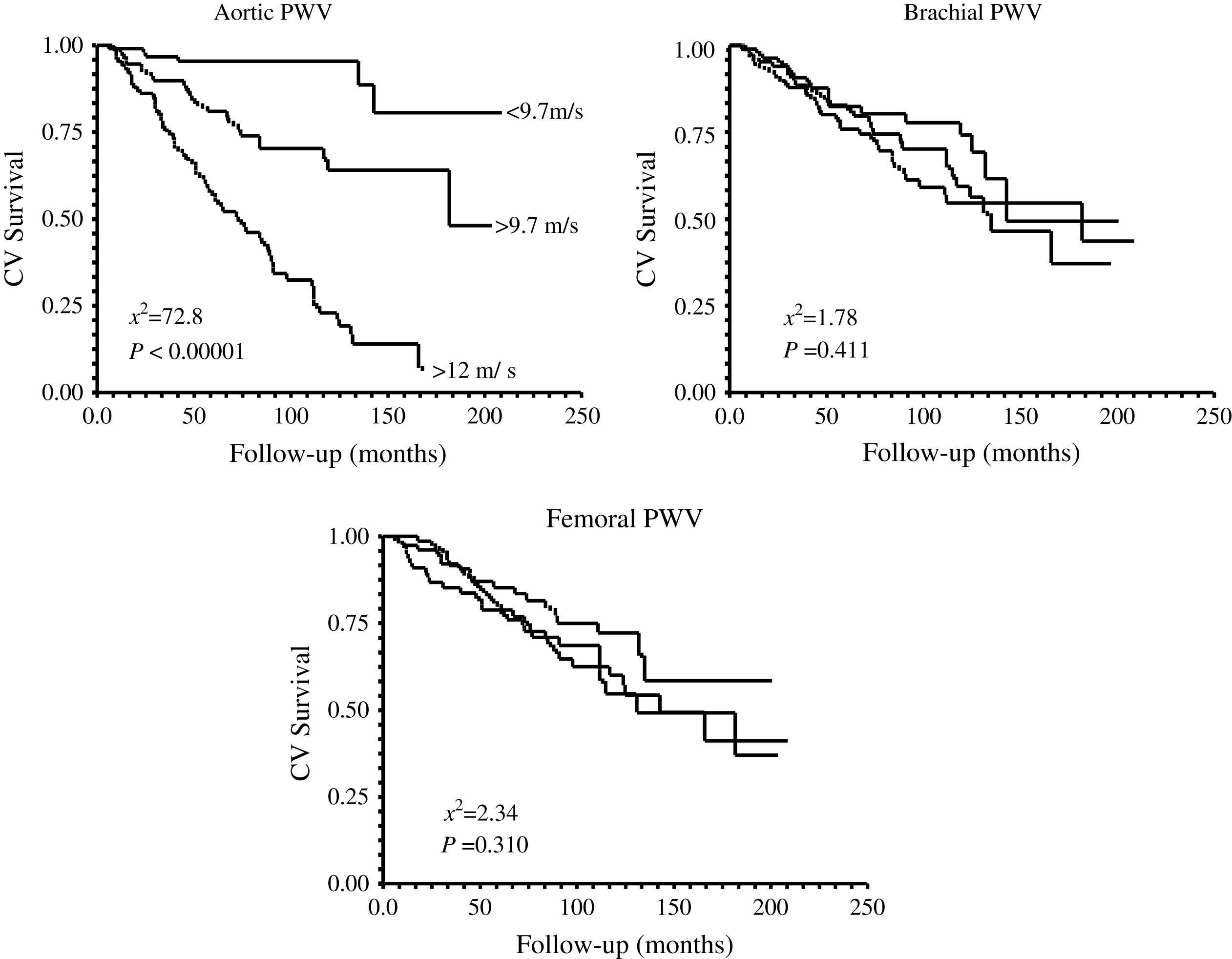
Kaplan–Meier survival curves for aortic, brachial, and femoral pulse waves velocities in end-stage renal disease patients.
In conclusion, the vascular complications in ESRD are ascribed to two different but associated mechanisms, namely atherosclerosis and arteriosclerosis. Arteriosclerosis characterized by diffuse dilation and hypertrophy of large conduit arteries and stiffening of arterial walls represents a clinical form of an accelerated aging process. These alterations are associated with several hemodynamic changes characteristic of ESRD, such as low shear stress and increased circumferential tensile stress due to increased arterial diameters and/or intra-arterial pressure. Resulting from these arterial changes, the systolic pressure is abnormally increased in ESRD patients while diastolic pressure is usually within the normal range or even low. The main adverse effects of arterial stiffening are: (1) an elevated LV afterload with development of LV hypertrophy and increased myocardial oxygen demand; and (2) altered coronary perfusion and blood flow distribution with relative subendocardial ischemia. Epidemiological studies have demonstrated the impact of arterial abnormalities on cardiovascular disease evolution and identified arterial remodeling and stiffening as independent predictors of overall and cardiac mortality in ESRD patients.
Acknowledgments
This work was supported by G.E.P.I.R. (Groupe d’Etude de Physiopathologie de l’Insuffisance Rénale) and INSERM U632.
References
Cite this article
TY - JOUR AU - Bruno Pannier AU - Alain P. Guérin AU - Sylvain J. Marchais AU - Fabien Métivier AU - Gérard M. London PY - 2007 DA - 2007/07/27 TI - Arterial structure and function in end-stage renal disease JO - Artery Research SP - 79 EP - 88 VL - 1 IS - 2 SN - 1876-4401 UR - https://doi.org/10.1016/j.artres.2007.06.001 DO - 10.1016/j.artres.2007.06.001 ID - Pannier2007 ER -