
Molecular determinants of arterial stiffness
- DOI
- 10.1016/j.artres.2007.03.004How to use a DOI?
- Keywords
- Hypertension; Monogenic disease; Extra-cellular matrix; Biomechanics; Gene profile
- Abstract
Arterial stiffness has an independent predictive value for cardiovascular events. This review proposes an integrated view of the molecular determinants of arterial stiffness, based on a candidate gene approach, an analysis of the structure–function relationship in hypertension, and studies on gene expression profile in humans. In monogenic diseases of connective tissue (Marfan, Williams, and Ehlers–Danlos syndromes) and corresponding animal models, the precise characterization of arterial phenotype allows understanding the influence of abnormal, genetically determined, wall components on arterial stiffness. These studies underline the role of extra-cellular matrix signaling in the vascular wall and the fact that elastin and collagen have not only passive elastic or rigid properties, but also are implicated in the control of SMC function. In animal models of essential hypertension (SHR and SHR-SP), the structural modifications of the arterial wall include a higher number of elastin/SMC connections, and smaller fenestrations of the internal elastic lamina, which could redistribute the mechanical load towards elastic materials. Thus, the changes in arterial wall material which accompany wall hypertrophy in these animals are not associated with an increased stiffness. Taken together, these data afford strong arguments to consider that arterial stiffness is not only influenced by the amount and density of stiff wall material, but mainly by its spatial organization.
- Copyright
- © 2007 Published by Elsevier B.V. on behalf of Association for Research into Arterial Structure and Physiology.
- Open Access
- This is an open access article distributed under the CC BY-NC license.
Large artery stiffness is the main determinant of pulse pressure (PP).1,2 Both have predictive value for cardiovascular events, independent of classical cardiovascular risk factors.3 Aortic stiffness has an independent predictive value for total and cardiovascular mortality, coronary morbidity–mortality, and fatal stroke, in patients with either essential hypertension, end-stage renal failure, or diabetes mellitus.3 This review proposes an integrated view of the molecular determinants of arterial stiffness, based on a candidate gene approach, an analysis of the structure–function relationship in hypertension, and studies on gene expression profile in humans.
Aging, hypertension, diabetes, chronic renal insufficiency and inflammatory disease
Aging and blood pressure are the 2 major determinants of arterial stiffness. Arterial stiffness is also increased in metabolic syndrome, type-1 and type-2 diabetes, chronic renal insufficiency and inflammatory diseases, which alter a large number of arterial wall components to various extents.
The normal media layer is composed of smooth muscle cells (SMC) and extra-cellular matrix (ECM). The 2 major scaffolding proteins, collagen and elastin, provide structural integrity and elasticity. Any alteration in collagen and elastin production and molecular repair mechanisms can theoretically change arterial elasticity.4 The relative content of collagen and elastin is controlled by a dynamic process of production and degradation. Degradation occurs through collagenases and elastases, produced by inflammatory cells such as macrophages and polymorphonuclear neutrophils, and gelatinase activation (MMP-2 and MMP-9).
With age, in the load-bearing media of elastic arteries, there is a loss of the orderly arrangement of elastic fibers and laminae, which display thinning, splitting, fraying and fragmentation.5 Degeneration of elastic fibers is associated with an increase in collagenous material and in ground substance and often with the deposition of calcium in this and in degenerate elastic fibers. In several pathological conditions, these changes are associated with an infiltration of vascular SMC, macrophages and mononuclear cells, and an increased content of matrix metalloproteinases and cytokines.6 Inflammation, either acute such as in systemic vasculitis or chronic during rheumatoid arthritis, may stiffen the large arteries through various mechanisms,7 including endothelial dysfunction, leading to “functional” stiffening of the large arteries (reduction of nitric oxide bioavailability and increased activity of opposing mediators such as endothelin-1), cell release of a number of inducible matrix metalloproteinases (including MMP-9), medial calcifications, changes in proteoglycan composition and state of hydration, and cellular infiltration around the vasa vasorum leading to vessel ischaemia.
Abnormal, stiffer collagen molecules can result from nonenzymatic glycation cross-linking, for instance during impaired glucose tolerance or diabetes. These irreversible cross-links form advanced glycation end products (AGEs).5 Not only in diabetes, but also in chronic renal diseases, does the activation of systemic and local renin–angiotensin system promote SMC proliferation, low-grade inflammation, and increase collagen content and AGE formation. Arterial stiffening in renal disease is also due to calcifications in the arterial media. Shanahan et al.8 have suggested a sequence of molecular events in vascular calcification beginning with the loss of expression by VSMCs, of constitutive inhibitory proteins, and ending with expression by VSMCs and macrophages of chondrocytic-, osteoblastic-, and osteoclastic-associated proteins that orchestrate the calcification process.
Thus, with aging, hypertension, diabetes, chronic renal disease and inflammation, the arterial wall can stiffen in response to a combination of several mechanisms. However, the respective role of each process is difficult to determine.
Candidate gene approach
A candidate gene approach may help to determine the pathogenic role of a given molecular determinant in arterial stiffening. This paragraph will review several studies concerning monogenic diseases of the arterial wall in humans and related animal models. They constitute interesting models for understanding the structure–function relationship of the arterial wall, particularly the influence of abnormal, genetically determined, wall components on arterial stiffness. Monogenic diseases of the arterial wall are complicated by major cardiovascular events, either dilatation, dissection and rupture (Marfan syndrome, Ehlers–Danlos syndrome) or hypertrophy and stenosis (Williams syndrome, Pseudoxanthoma Elasticum9).
Marfan syndrome
The Marfan syndrome (MFS) is a connective tissue disorder inherited as an autosomal dominant trait, characterized by abnormalities involving the skeletal, ocular, and cardiovascular systems.10 MFS results from mutations in the gene encoding fibrillin-1 (FBN1) and subsequent abnormalities in elastic fibers assembly.10 In MFS patients, the increased arterial stiffness was confined to the aorta, with no difference at the site of the carotid, femoral and radial arteries.11
A clinical hallmark and the major cause of morbidity and premature death in MFS is aortic root dilation and associated aortic regurgitation, dissection and rupture.10 The exact mechanisms leading to dilatation are not fully understood, but steady and pulsatile stresses are likely to play an important role through mechanical fatigue of abnormal elastic fibers and microdissections.10 Indeed, in MFS patients, central PP, which is influenced by aortic stiffness, was a powerful determinant of ascending aorta diameter, independently of age and body surface area, whereas mean BP was not.11
In a rodent model of the Marfan syndrome underexpressing FBN1, aortic wall stiffness was 4-fold increased compared to wild-type strains.12 Electron microscopy showed an unusually smooth surface of elastic laminae, manifesting the loss of cell attachments that are normally mediated by fibrillin-1.13 Bunton et al.13 suggested that the loss of cell attachments signals a nonproductive program to synthesize and remodel an elastic matrix. Thus, fibrillin-1 appears as a key element for a normal spatial organization of the arterial wall, adequately loading the elastic components thus maintaining a physiological arterial stiffness.
Similar findings were observed in mice lacking desmin.14 In k.o. mice, arterial SMC have lost part of their connections to the extra-cellular matrix, particularly SMC fingerlike-projections to elastic lamellae were reduced, and carotid stiffness was higher than in wild mice.14 In addition, mice lacking desmin showed a reduction of in vitro breaking pressure. Since the solidity of the arterial wall was decreased in the Marfan syndrome and mice lacking desmin, connections between ECM and VSMC are very likely involved both in arterial elasticity and in mechanical strength.
Williams syndrome
Williams syndrome (WS) is a genetic disorder characterized by mental and statural deficiency, and cardiovascular abnormalities including peripheral arterial stenoses and hypertension.15 These features may be related to the deletion of one allele of the elastin gene.15 Indeed, in mice lacking elastin, the absence of elastin was sufficient to induce subendothelial proliferation of smooth muscle cells and contribute to obstructive arterial disease. Thus, elastin has not a purely structural role, but also a regulatory function during arterial development, controlling proliferation of smooth muscle and stabilizing arterial structure.16 In patients with Williams syndrome, the carotid artery was abnormally distensible and thick, and electron microscopy of renal artery stenosis material showed SMC dedifferentiation.17 We suggest that SMC dedifferentiation, leading to arterial wall hypertrophy, may represent a major factor responsible for increased distensibility.
Ehlers–Danlos syndrome
Ehlers–Danlos syndrome (EDS) type-IV, the vascular type (vEDS), which results from mutations in the gene for type-III procollagen,18 is characterized mainly by spontaneous rupture of arteries.18 In a recent cross-sectional study,19 we did not observe any change in arterial stiffness, but an abnormally thin carotid artery, leading to a higher circumferential wall stress, which in turn may explain the occurrence of dissection and rupture of a fragile arterial tissue. The fragility of the arterial wall in mice with mutations in type-I and type-III collagens20 was attributed to the dramatic reduction of the collagen type-I fibrils in the aortic media and adventitia.
Thoracic aortic aneurysm and/or aortic dissection and patent ductus arteriosus
I was recently demonstrated that a genetic disease combining thoracic aortic aneurysm and/or aortic dissection and patent ductus arteriosus was caused by mutations in the MYH11 gene.21 These mutations alter the C-terminal coiled-coil region of the smooth muscle myosin heavy chain, a specific contractile protein of SMC. All individuals bearing the heterozygous mutations, even if asymptomatic, showed marked aortic stiffness. Examination of pathological aortas showed large areas of medial degeneration with very low SMC content. Thus, MYH11 heterozygous mutation leads to an early and severe decrease in the elasticity of the aortic wall, consistent with the role of SMC in maintaining the mechanical properties of the thoracic aorta.22
In conclusion, the precise characterization of arterial phenotype in monogenic diseases of connective tissue allows understanding the influence of abnormal, genetically determined, wall components on arterial stiffness. In addition, these data underline the role of ECM signaling in the vascular wall and the fact that elastin and collagen have not only passive elastic or rigid properties, but also are implicated in the control of SMC function: migration, proliferation, adhesion and cytoskeletal rearrangement.22
Integrative physiology of arterial stiffness in essential hypertension
Animal models of essential hypertension, such as spontaneously hypertensive rats (SHR) and stroke prone-SHR (SHR-SP) can provide new insights into the cellular and molecular determinants of arterial stiffness. During essential hypertension, the sustained increase in blood pressure is a trigger for the development of arterial wall hypertrophy, which in turns allows normalizing circumferential wall stress (σθ), according to Lame’s equation (σθ = MBP.R/h, where R is the radius and h is the wall thickness). According to physics laws, any increase in wall thickness should increase arterial stiffness for a given BP level, because of the juxtaposition of material having identical mechanical properties. Surprisingly, we and others found that hypertrophy, observed in HT, was accompanied by a reduced stiffness of the wall material (Young’s elastic modulus), and a normal stiffness of the artery considered as a whole.23,24 Similar findings were observed in SHR and SHR-SP at the site of the carotid artery and the abdominal aorta, when hypertensive strains were compared to Wistar–Kyoto rats.25,26 Therefore, the hypertension-induced wall thickening was not associated with an increased stiffness in patients with essential hypertension and rat models of hypertension.
We hypothesized that sustained hypertension was associated with a rearrangement of the arterial wall material, implying qualitative or quantitative changes in arterial components leading to the mechanical adaptation of the arterial wall to higher levels of BP. Indeed, the 3-D organization of stiff elements may play a more important role than their content and density in the global mechanical behavior of the artery. To further investigate the role of wall components in the chronic adaptation of the arterial wall to high levels of BP, we focused on fibronectin, dense plaques, and fenestrations. Fibronectin (FN) plays an important role in cell–matrix interactions, through an interaction with specific cellular integrin receptors, such as the α5β1-integrin. We found an increase in total FN, cellular FN (EIIIA FN isoform), and α5-integrin in the aortas of SHRs25 and SHR-SP.26 Dense plaques of smooth muscle are a major site of anchorage between SMC and extra-cellular matrix. We demonstrated by electron microscopy27 that the percentage of cell surface occupied by dense plaques and connected to the elastic lamellae in the aorta was twice as high in SHR compared with Wistar rats. Thus, the elastin network may contribute to the mechanical adaptation of the arterial wall in SHR, not only through variations of its total amount but through variations of the extent of anchorage to the muscle cells. This anchorage, which concerns not only the internal elastic lamina but also the elastic lamellae participating to the musculo-elastic fascicles described by Wolinsky and Glagov,28 may thus play a role both in medium-sized muscular arteries and in large elastic arteries. Mechanical stress is concentrated close to the fenestrations of elastic lamellae. Using confocal microscopy, we showed26 that the mean area of fenestrations and fraction of area occupied by fenestrations of the internal elastic lamina (IEL) were smaller in SHR-SP and SHR than in WKY. Thus, a reduction in the stress-concentration effects within the IEL may represent an adaptive mechanism to protect the elastin network against an increased mean wall stress.
Fig. 1 summarizes the structural modifications of the arterial wall in SHR and SHR-SP, which could play a role in the mechanical adaptation to a high level of wall stress. These changes, which include a higher number of FN/α5β1-integrin complexes and elastin/SMC connections, and smaller IEL fenestrations, could redistribute the mechanical load towards elastic materials. They provide mechanisms explaining that the changes in arterial wall material which accompany wall hypertrophy in animal models of essential hypertension are not associated with an increased stiffness and mechanical strength.
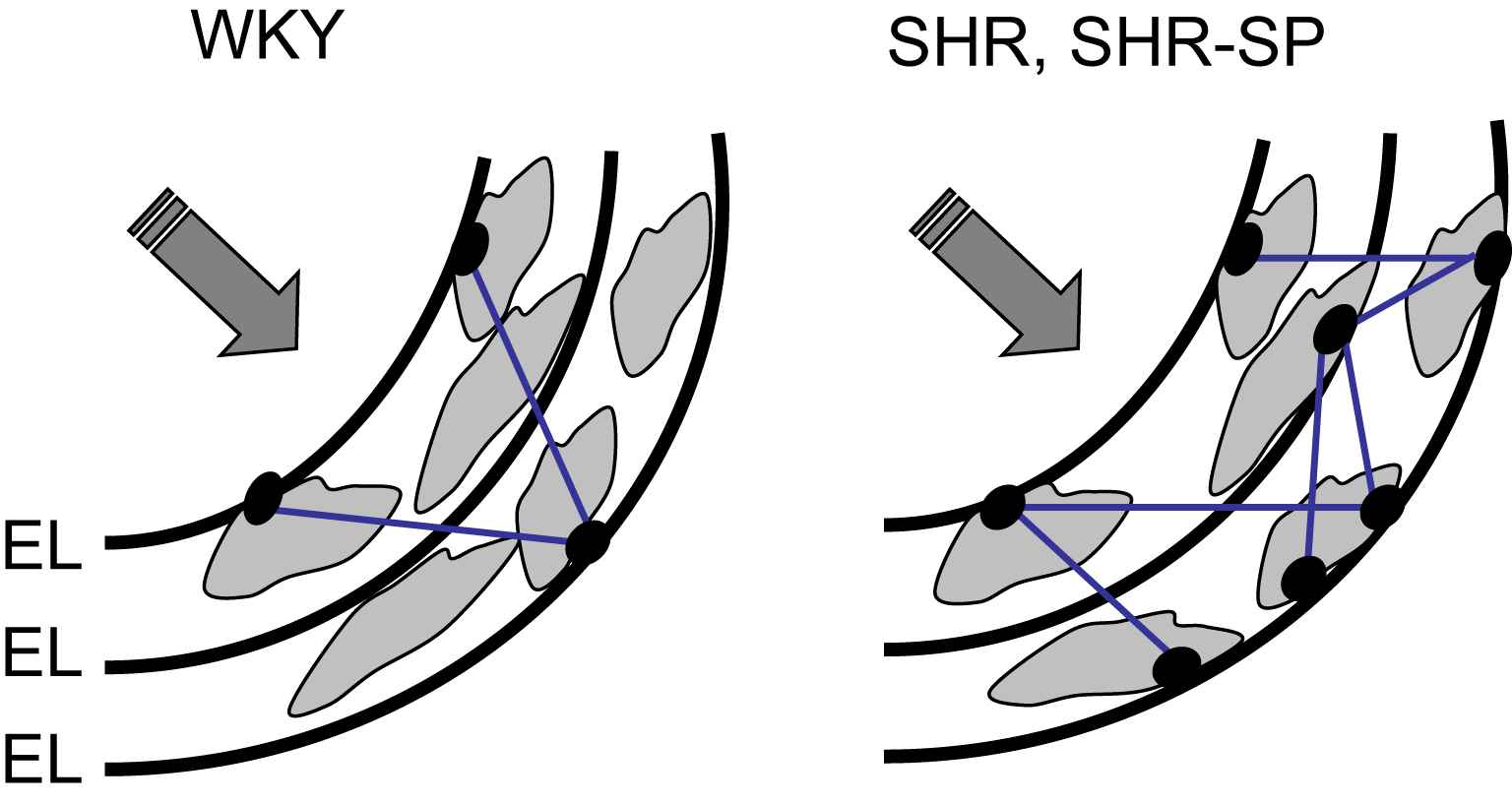
Increased loading of elastic materials in rat models of essential hypertension (with permission of Ref. 22). The structural modifications of the arterial wall in SHR and SHR-SP, which include a higher number of connections between smooth muscle cells (SMC) and the internal elastic lamina (IEL) through dense plaques (black dots), and smaller IEL fenestrations, could redistribute the mechanical load towards elastic materials. EL = elastic lamellae.
Gene expression profile of the aorta in hypertension
Given the importance of the 3-D organization of the arterial wall components in arterial stiffness and adaptive mechanisms in polygenic and monogenic diseases, we used a global approach to clarify the relationship between individual gene expression and arterial stiffness in humans. We used a microarray technology29 to identify novel transcriptional biomarkers of arterial stiffening. Our working hypothesis was that patients with abnormal aortic stiffness had a specific gene expression profile in aortic wall tissue (genes abnormally over- or under-expressed), compared to genes expressed in patients having a normal arterial stiffness. Aortic biopsies originated from patients with coronary heart disease undergoing a coronary artery bypass graft. RNA samples were analysed using the Affymetrix GeneChip® technology. On the 12,558 transcripts, 151 probe sets were differentially expressed between stiff and distensible aortas, among which 32 were significantly associated with PWV, through a positive or negative correlation. The majority were related to cytoskeleton, with the remainder distributed between matrix and membrane. Differentially expressed genes, which were involved in the mechanical regulation of vascular structure, included integrins (α2b, α6, β3 and β5), proteoglycans (decorin, osteomodulin…), fibulin-1, and fascin. Nearly half of abnormal transcripts could be classified as signaling/communication or cell structure/motility30 (Fig. 2A). We identified 2 distinct groups of genes,29 those associated with cell signaling and those associated with the mechanical regulation of vascular structure (cytoskeletal, cell membrane, extra-cellular matrix).

Repeatability of gene expression profile in human aorta. (A) Affymetrix GeneChip® technology, using 12,558 transcripts: 151 probe sets were differentially expressed between stiff and distensible aortas. (B) CNRS Gene chips containing 27,648 oligonucleotides probes specific to 25,279 human transcripts: 65 probe sets were differentially expressed between stiff and distensible aortas. Genes classified as signaling/communication or cell structure/motility represented the most important group (39 and 43% in Fig. 2A, B, respectively). (C) The fold-changes of the 28 genes significantly associated with PWV and present on the 2 different chips were comparable between the 2 methods (open column: Affymetrix GeneChip®; grey column: CNRS Gene chip). TBCE: tubulin-specific chaperone E; CTNND: catenin delta 1; ITGAB: integrin, α2b; DGCR6: DiGeorge syndrome critical region gene 6; AGC1: aggrecan 1; MYO1C: myosin IB; SSH3BP1: spectrin SH3 domain binding protein 1; ITGB3: integrin β3; CKAP4: cytoskeleton-associated protein 4, transmembrane protein, endoplasmic reticulum/Golgi intermediate compartment; EPB41L2: erythrocyte membrane protein band 4.1-like 2; ACTG1: actin, gamma 1; CSPG5: chondroitin sulfate proteoglycan 5 (neuroglycan C); GABARAP: ganglioside expression factor 2; MFAP2: microfibrillar-associated protein 2; and PRPH: peripherin.
Transcriptome analysis is often criticized for a lack of repeatability. To confirm these results, we performed a second experiment in 9 different patients. RNA samples were analysed using arrays containing 27,648 oligonucleotides probes specific to 25,279 human transcripts (supplied by CNRS UMR 6097, Sophia-Antipolis). On the 27,648 probes, we isolated 65 genes differentially expressed between stiff and distensible aortas, following the same analysis process. Among these 65 transcripts, genes classified as signaling/communication or cell structure/motility represented the most important group [26 and 17%, respectively, i.e. 43% (Fig. 2B), an amount similar to the 39% observed in the first experiment (Fig. 2A)], demonstrating the repeatability of the results with 2 different techniques of microarray. Moreover, the fold-changes of the 28 genes significantly associated with PWV and present on the 2 different chips were comparable between the 2 methods (Fig. 2C).
Although studies previously discussed in this review have concentrated on the contribution of the ECM towards arterial stiffness, these data suggest that changes in expression of signaling molecules play an equally very important role. Alterations in the profiles of signaling molecules could be involved in the regulation of cell cytoskeletal organization, cell–matrix interactions, or the contractile state of the cell.
Therapeutic applications
The identification of genes and proteins differentially expressed in normal and pathological aortic tissue would greatly aid to the determination of potential pharmacological targets to reduce arterial stiffness when BP is optimally reduced. The most interesting targets for drug candidates should theoretically fulfil the following conditions: (a) these proteins are encoded by genes found to be over- or under-expressed in stiff aortas; (b) these proteins are found to be over- or under-expressed in aortic tissue; (c) if there were a corresponding genetic polymorphism, the genotype–phenotype analysis should have observed an abnormally high or low arterial stiffness in carriers of the specific polymorphism; (d) if there were a corresponding monogenic disease, the genotype–phenotype analysis should have observed an abnormally high or low arterial stiffness in these patients. Candidate proteins could theoretically be either an already known target corresponding to an existing drug (collagen I, elastin), an identified target without existing drug (fibrillin-1, fibulin), or a target never identified for its role in arterial stiffness (decorin, osteomodulin).
Conclusion
Recently, animal and human studies have further investigated the molecular bases of arterial stiffness. They have shown that several genes and molecules are associated with vascular wall stiffening and illustrated the consequences of their changes in various clinical conditions. These molecules and their signaling remain to be elucidated for future drug treatments of arterial stiffness.
References
Cite this article
TY - JOUR AU - Stéphane Laurent AU - Céline Fassot AU - Patrick Lacolley AU - Pierre Boutouyrie PY - 2007 DA - 2007/06/06 TI - Molecular determinants of arterial stiffness JO - Artery Research SP - 26 EP - 31 VL - 1 IS - 1 SN - 1876-4401 UR - https://doi.org/10.1016/j.artres.2007.03.004 DO - 10.1016/j.artres.2007.03.004 ID - Laurent2007 ER -