
The reality of aging viewed from the arterial wall☆
McDonald Lecture, ARTERY 12, Vienna, Austria, October 20, 2012.
- DOI
- 10.1016/j.artres.2013.01.003How to use a DOI?
- Copyright
- © 2013 Published by Elsevier B.V. on behalf of Association for Research into Arterial Structure and Physiology.
- Open Access
- This is an open access article distributed under the CC BY-NC license.
Any discussion about any aspect of aging cannot beg the issue of what aging is. This is a tough question, and there are numerous perspectives regarding the answer. My view is that aging is “a shift in an organism’s reality.” So what’s reality? This is another tough question. My view is that reality can be comprehensively defined as a system of mutual enslavement of DNA and its environment. If this appears to be a naïve assessment of reality, check out what constitutes the DNA environment (Fig. 1).
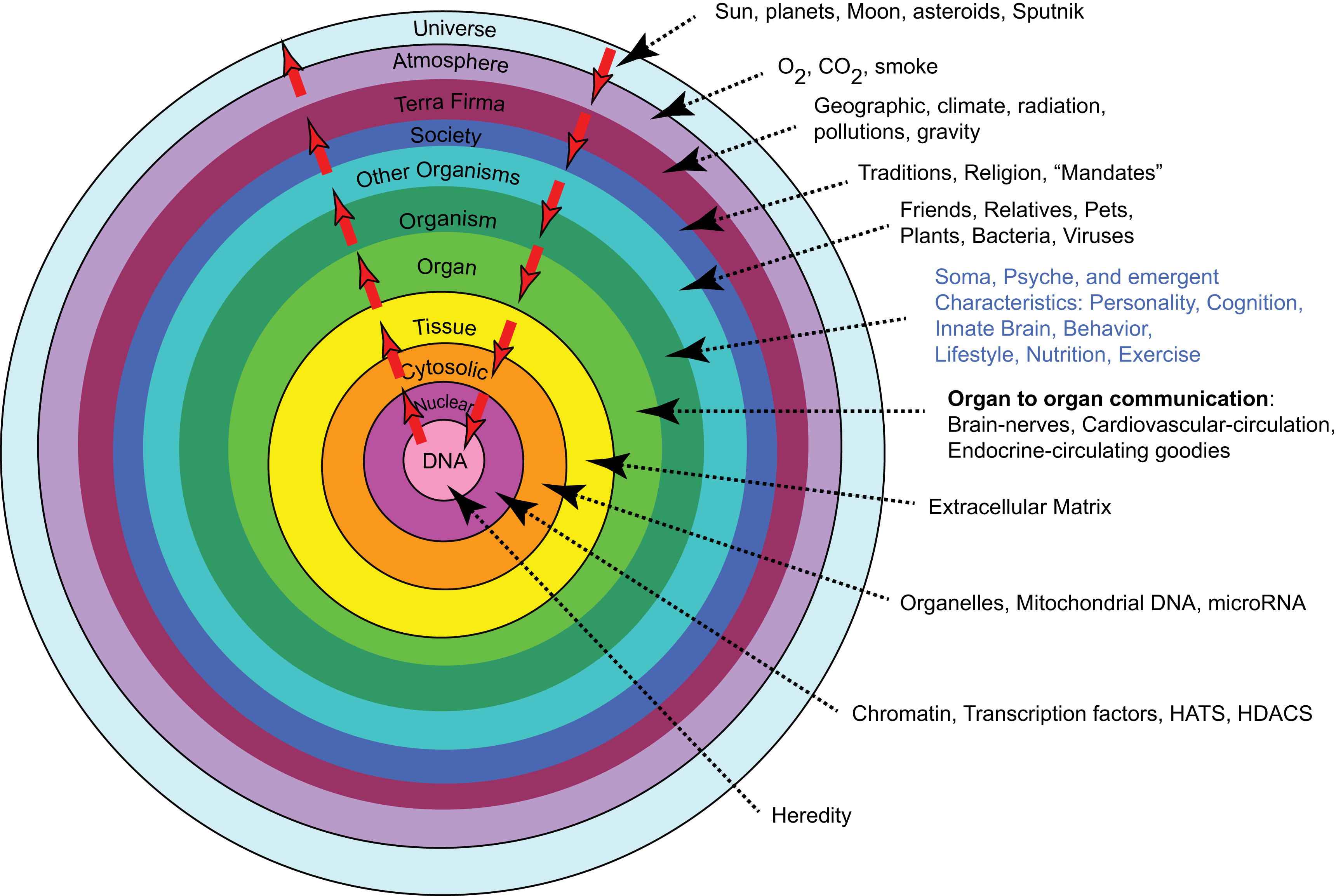
Reality is a system of mutually enslaved DNA and its environment.
The white arrows in the figure indicate continual bidirectional signaling that must occur to sustain our existence. In this context, aging can be construed as a series of failures of the signaling within the DNA environmental system. A general description of some failing interactions in this system includes: generation of signals, sensing of signals, signal transmission, response to signals, and proteostasis.
Viewing the reality of aging from the arterial wall begins with the realization that arterial diseases, e.g. atherosclerosis and hypertension, are rampant in Western society, and increase exponentially with advancing age. Progressive changes occur throughout life in the structure and function of central arteries in numerous species. These changes include diffuse intimal and medial thickening, and enhanced stiffening.1 Since the likelihood for predominantly systolic hypertension and atherosclerosis to occur increases in epidemic proportion among older persons1,2 (Fig. 2), it is reasonable to hypothesize that specific mechanisms that underlie alterations in the arterial substrate that accompany “aging” may be intimately linked to the age-associated exponential increase in predominantly systolic hypertension.1 Indeed, recent studies (Fig. 3) show pulse wave velocity, an index of arterial stiffness, is an independent predictor of the future increases in SBP and of incident hypertension.3
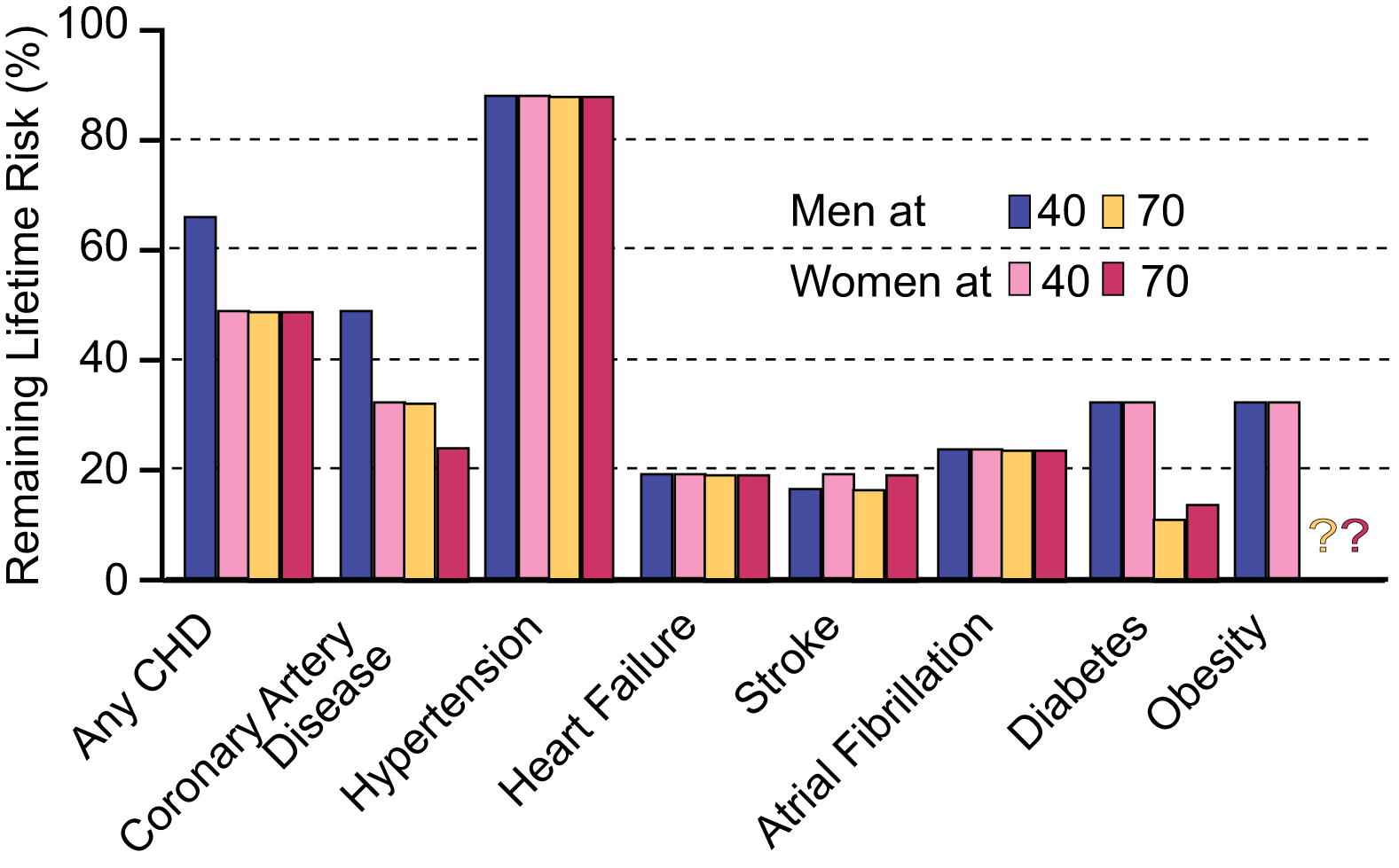
Remaining lifetime risk for CVD and other diseases among men and women free of disease at 40 and 70 years of age.
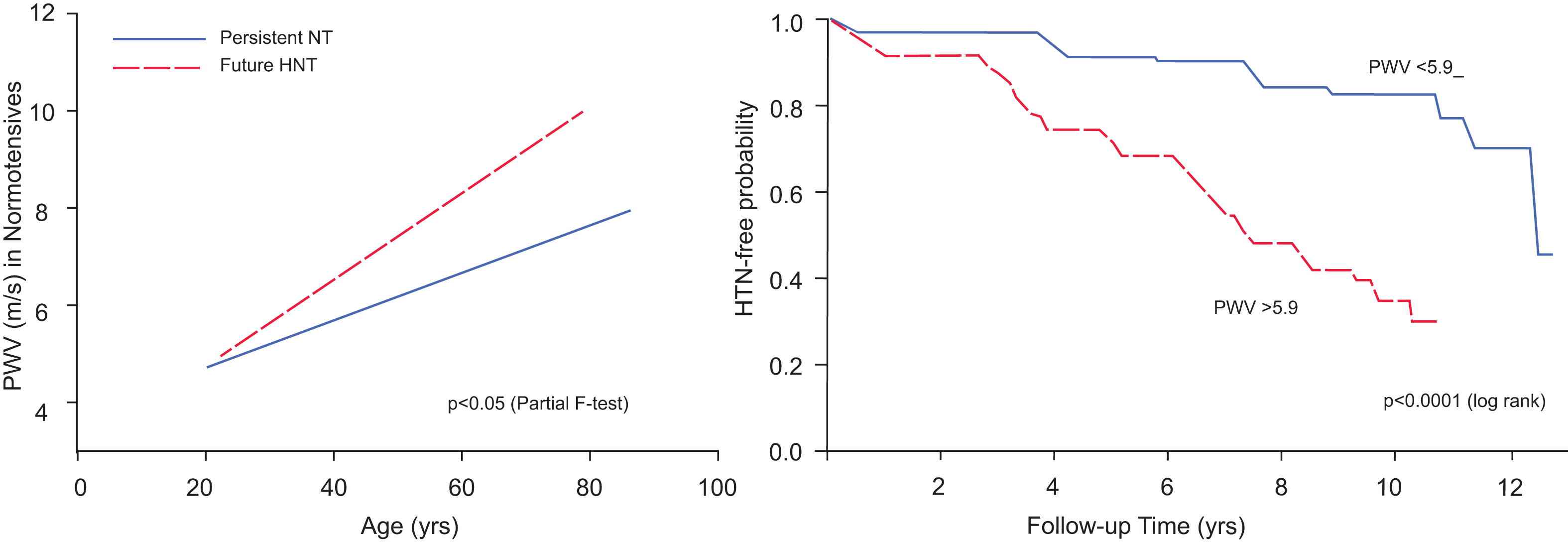
PWV predicts future blood pressure rise and development of hypertension.
Age-associated remodeling of the aortic wall of both animals and humans involves a proinflammatory profile of arterial cell and matrix properties4 (Fig. 4).

Aging of large arteries: inflammation.
This profile features increased production of angiotensin II (Ang II) and increased vascular smooth muscle cell expression and secretion of downstream Ang II/AT1, mineralocorticoid and endothelin receptor signaling molecules (Fig. 5), e.g., matrix metalloproteases (MMPs), calpain-1 and monocyte chemoattractant protein (MCP-1), transforming growth factor β1 (TGF-β1) NFκb, TNFα, iNOS, and VCAM. Activation of calpain-1, MMPs, TGF-β, and NADPH oxidase within the arterial wall is increased, and nitric oxide bioavailability is reduced.1,3,5–7 Invasive, proliferative and secretory capacities of early passage vascular smooth muscle cells (VSMC) isolated from the aged arterial wall are increased, and are linked to augmented Ang II signaling. This age-associated arterial proinflammatory secretory profile within the grossly appearing arterial wall and related structural/functional remodeling of cells and matrix is reproduced in young rats by chronic infusion of Ang-II.5
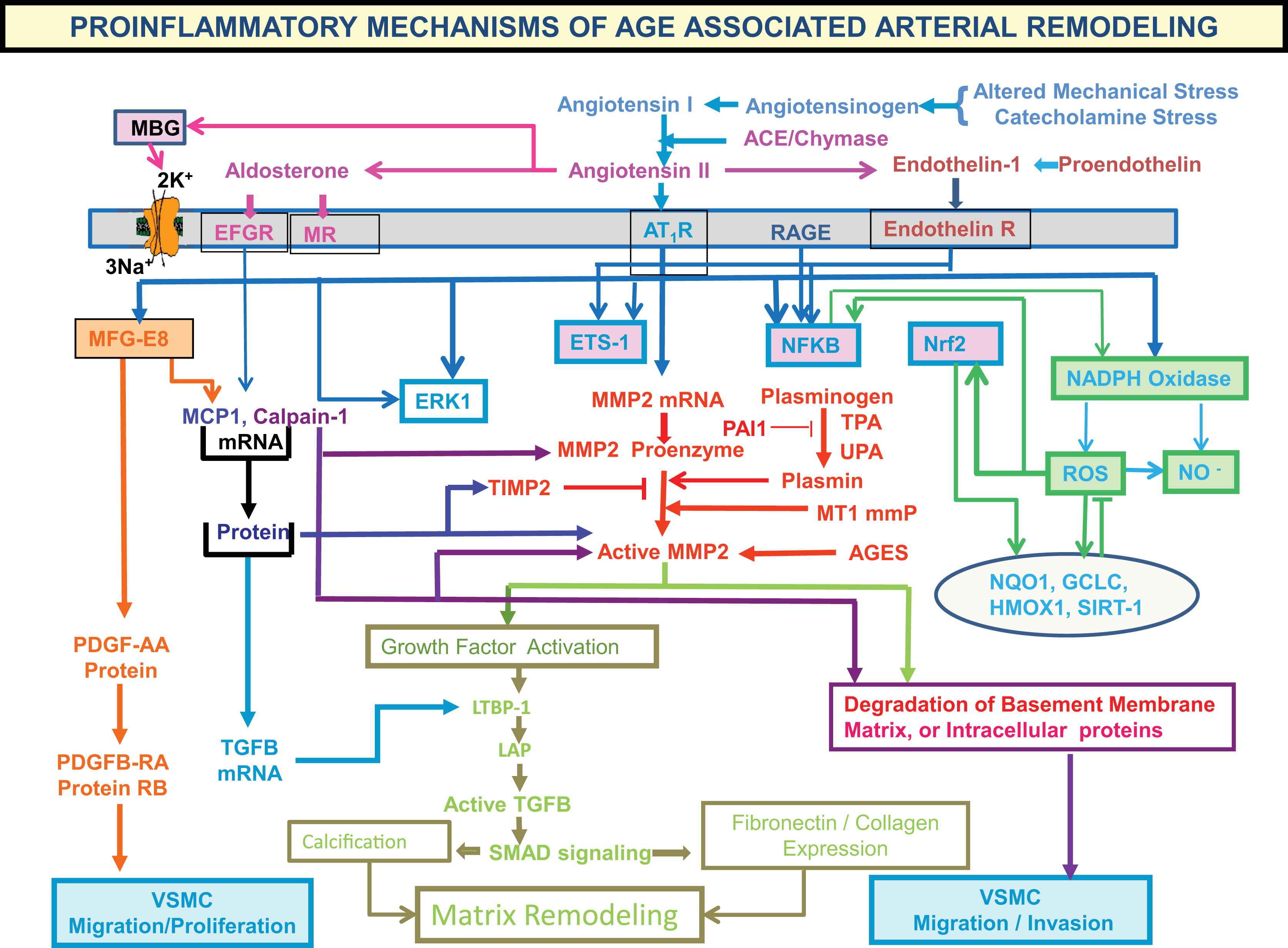
Proinflammatory mechanisms of age-associated arterial remodeling.
Milk fat globule protein E8
But the aortic wall thickening induced by aging likely results from the concerted effects of numerous signaling proteins that have yet to be identified. A comprehensive quantitative proteomic study to analyze aortic proteins from young (8 months) and old (30 months) rats and identified 50 proteins that significantly change in abundance with aging.8 The expression of one novel protein, the milk fat globule protein epidermal growth factor 8 (MFG-E8), increases 2.3-fold in abundance in old aorta. Transcription and translation analysis demonstrated that aortic MFG-E8 mRNA and protein levels increase with aging in several mammalian species including humans. An accumulation of milk fat globule EGF-8 protein (MFG-E8) occurs within the context of arterial wall inflammatory remodeling in hypertension, diabetes mellitus, or atherosclerosis. Dual immunolabeling shows that MFG-E8 colocalizes with both angiotensin II and monocyte chemoattractant protein (MCP)-1 within vascular smooth muscle cells (VSMCs) of the thickened aged aortic wall. Exposure of early passage VSMCs from young aorta to angiotensin II markedly increases MFG-E8 and enhances invasive capacity to levels observed in VSMCs from old rats. MFG-E8 colocalizes with both angiotensin II and MCP-1 within VSMCs of the thickened aged aortic wall. Treatment of VSMCs with MFG-E8 increases MCP-1 expression and VSMCs invasion, which are inhibited by the MCP-1 receptor blocker vCCI. Silencing MFG-E8 RNA substantially reduces MFG-E8 expression and VSMCs invasion capacity.
MFG-E8 not only induces VSMC invasion, but also affects VSMC proliferation, which is a salient feature of arterial inflammation. PCNA and Ki67, markers of cell cycle activation, increase in the rat arterial wall in vivo during aging between 8 and 30 months.9 In fresh and early passage VSMC isolated from old aortae, an increase in CDK4 and PCNA, an increase in the acceleration of cell cycle S and G2 phases, decrease in the G1/G0 phase, and an increase in PDGF and its receptors confer elevated proliferative capacity, compared to young VSMC. Increased coexpression and physical interaction of MFG-E8 and integrin αvβ5, the cell surface receptors for MFG-E8, occur with aging in both the rat aortic wall in vivo and in VSMC in vitro. In young VSMC in vitro, MFG-E8 added exogenously, or overexpressed endogenously, triggers phosphorylation of ERK1/2, augments levels of PCNA and CDK4, increases BrdU incorporation, and promotes proliferation, via αvβ5 integrins. MFG-E8 silencing, or its receptor inhibition, or blockade of ERK1/2 phosphorylation in these cells reduces PCNA and CDK4 levels and decelerates the cell cycle S phase, conferring a reduction in proliferative capacity. Collectively, these results indicate that MFG-E8 in a dose-dependent manner coordinates the expression of cell cycle molecules and facilitates VSMC proliferation via integrin/ERK1/2 signaling, indicating that enhanced MFG-E8 signaling is a mechanism of the age-associated increase in aortic VSMC proliferation. Chronic infusion of Ang II into young rats increases aortic MFG-E8, MCP-1 and PCNA, an index of cellular proliferation, to levels in untreated old rats. (Fig. 6).

MFG-E8 stimulates proliferation and invasion in VSMC, and its medin degradation product may play a role in increased arterial stiffness.
Medin amyloid
Characteristic features and pathophysiology of amyloid proteins
Misfolding of extracellular protein to form amyloid deposits is a dynamic process, occurring in parallel with, or as an alternative to physiologic folding, generates insoluble protein aggregates that are deposited in tissues.10 The incidence of aortic amyloidosis averaged 79% in 224 autopsy cases. The incidence prior to the 5th decade was 51% and it rose sharply with age and reached 95% in over the 8th decade. The incidence of aortic amyloidosis was always higher than in the heart, in which amyloidosis also increased with age. The aortic media exhibited the majority of amyloid content, which consisted of numerous minute deposits without a relationship to atheromata.11
Specific amyloid types are defined, on the basis of its assembled protein fibril patterns. Initially, localized amyloid deposits, limited to certain organs or tissues, were regarded as innocent bystanders, or by-products of diseases, rather than having involvement in their pathogenesis. This view has changed radically during the last decade.12 Small amyloid deposits, or oligomeric preamyloid aggregates of specific amyloid fibril proteins, are now believed to be critical factors toxic cellular effects involved in the pathogenesis of common disorders, e.g. the amyloid b-peptide (Ab) in Alzheimer’s disease and islet amyloid polypeptide (IAPP) in type II diabetes.12 Although mechanisms by which protein aggregates lead to cell injury and death are poorly understood, and fibrils are potential cytotoxins,13 β sheet peptides (e.g., amyloid β) are known to form ion channels in lipid bilayers possibly through aggregation, though the channel structure is not clear.14
Medin amyloid (AMed)
Analysis of proteins extracted from amyloid-rich aortic media detected an amyloid protein called medin.15 Milk fat globule protein E8 (formerly called lactadherin or SED1) is the precursor protein of Medin amyloid, which becomes deposited in the aortic media in almost 100% of the Caucasian population over 50 years of age.16 Medin amyloid (AMed) is not restricted to the aorta and the temporal artery, but also occurs in other arteries, mainly in the upper part of the body, including intracranial vessels.15 Medin amyloid deposits also contain its parent molecule, MFGE8. The medin fragment is 5.5 kDa and derives from the C2-like domain of MFGE8.16 The C2-like domain has been shown to bind phosphatidylserine and the RGD motif binds αvβ3 and αvβ5 integrins.16 In vitro, medin forms amyloid-like fibrils and the last 18–19 medin amino acid residues are the aggregation-prone region.17 The two C-terminal phenylalanines may also favor amyloid formation.17
In human aortae, medin amyloid co-localizes with elastic fibers of arteries18 and is also associated with other elastic structures.16 There is some evidence to indicate that non-amyloid prefibrillar medin oligomeric aggregates may also be toxic to the surrounding cells. in vitro aggregated medin induces death of aortic smooth muscle cells and cells incubated together with medin increased the production of metalloproteinase-2, i.e. a protease that degrades elastin and collagen matrix metalloproteinase-2.15
Elastic fibers become arranged and anchored by a scaffold protein, Fibulin-5. Fibulin-5−/− mice display aortic abnormalities due to disorganized and fragmented elastic fibers.19,20 Since MFGE8 promotes RGD motif-dependent cell adhesion21 and binds to elastin, has been suggested that MFGE8, like fibulin-5,19 it is involved in organizing elastic fibers to cells.16 Both medin and MFGE8 bind to tropoelastin in a concentration-dependent fashion. It has been suggested that the medin domain mediates the MFGE8-tropoelastin interaction is a cell adhesion protein and its medin domain may connect smooth muscle cells to the elastic fibers of arteries.16 Given that both medin and MFGE8 interact with elastic fibers, elastin may be an important component in the formation of medin amyloid.16 It is believed medin may be a factor involved in the increased aortic stiffness that accompanies advancing age.16,18,22 Indeed, correlations between serum MFG-E8 and pulse wave velocity and cardiovascular risk factors have been observed older normal subjects and in elderly patients with type 2 diabetes mellitus.23
MFG-E8 and pathologic angiogenesis
Expression and secretion of glycosylated MFG-E8 in and around blood vessels plays a crucial role in vascular endothelial growth factor (VEGF)-dependent neovascularization in the adult mouse.24 By binding to its receptors, MFGE8 interacts with αvβ3 and αvβ5 integrins and alters both VEGF-dependent Akt phosphorylation and neovascularization in endothelial cells Thus, MFGE8 is an important target for the modulation of neovascularization.24 Recent evidence indicates that MFG-E8 is implicated in pericyte/pericyte precursor function and that pericyte-derived MFG-E8 regulates pathologic angiogenesis.25 In response to an increase in PDGF-BB, MFG-E8 binding to its integrin receptors associates with the PDGF receptors and focal adhesion kinase receptors and delays PDGF receptor degradation, serving to potentiate downstream PDGF signaling.26 These cell autonomous actions of MFG-E8 on pericytes or pericyte precursors is a potential mechanism of how MFG-E8 promotes angiogenesis.26
Thus the age-associated increase in MFG-E8 is a novel pivotal relay element within the angiotensin II/MCP-1/ERK, CDK4 VSMC invasion and proliferation signaling cascades, and medin amyloid production. Targeting of MFG-E8 within this signaling axis pathway (Fig. 5) is a potential novel therapy to reduce or delay age-associated arterial stiffening and inflammation that are also featured in diseases that become rampant at older ages, such as atherosclerosis and hypertension.
Age-associated matrix remodeling of central arteries
In addition to amyloid deposition, central arterial fibrosis and calcification are features of age-associated arterial extracellular remodeling and also linked to Ang II signaling. Angiotensin II induces matrix metalloproteinase 2 (MMP2) and calpain-1 expression and activity in the arterial wall (Figs. 5 and 7).27,28 Dual histo-immunolabeling demonstrates co-localization of calpain-1 and MMP2 within old rat vascular smooth muscle cells.28 Over-expression of calpain-1 induces MMP2 transcripts, protein levels and activity, in part, by increasing the ratio of membrane-type 1 MMPs to tissue inhibitor of metalloproteinases 2. These effects of calpain-1 over-expression-induced MMP2 activation are linked to increased collagen I and III production and vascular calcification (Fig. 7).

Age-associated matrix remodeling of central arteries.
In addition, over-expression of calpain-1 also induces transforming growth factor-beta 1/Smad signaling, elastin degradation, alkaline phosphatase activation and total calcium content, but reduces the expression of calcification inhibitors, osteopontin and osteonectin, in cultured vascular smooth muscle cells in vitro and in carotid artery rings ex vivo. Furthermore, both calpain-1 and collagen II increase with aging within human aortic intima. Interestingly, in aged human aortic wall, both calpain-1 and collagen II are highly expressed in arteriosclerotic plaque areas compared to grossly normal areas. Cross-talk of two proteases, calpain-1 and MMP2, leads to secretion of active MMP2, which modulates ECM remodeling via enhancing collagen production and facilitating vascular calcification (Fig. 7). Thus, calpain-1 is a novel molecular target for retarding age-associated ECM remodeling and its attendant risk for hypertension and atherosclerosis.
Age-associated central arterial remodeling involving arterial wall collagen deposition and elastin fragmentation are linked to the age-associated increase in arterial pressure. As noted (Figs. 4 and 5), this arterial remodeling is linked to proinflammatory signaling, including transforming growth factor-β1, monocyte chemoattractant protein 1, and proendothelin 1, activated by extracellular matrix metalloproteinases (MMPs) and orchestrated, in part, by the transcriptional factor ets-1 (Fig. 8). Recent studies demonstrate that inhibition of MMP activation can decelerate the age-associated arterial proinflammation.29 Chronic administration of a broad-spectrum MMP inhibitor, PD166739, via a daily gavage, to 16-month-old rats for 8 months resulted in inhibition of the age-associated increases in aortic gelatinase and interstitial collagenase activity in situ; preservation of the elastic fiber network integrity; a reduction of collagen deposition; a reduction of monocyte chemoattractant protein 1 and transforming growth factor-β1 activation; a diminution in the activity of the profibrogenic signaling molecule SMAD-2/3 phosphorylation6; inhibition of proendothelin 1 activation7; downregulation of expression of ets-1; and marked blunting of the expected age-associated increases in arterial pressure (Fig. 9).
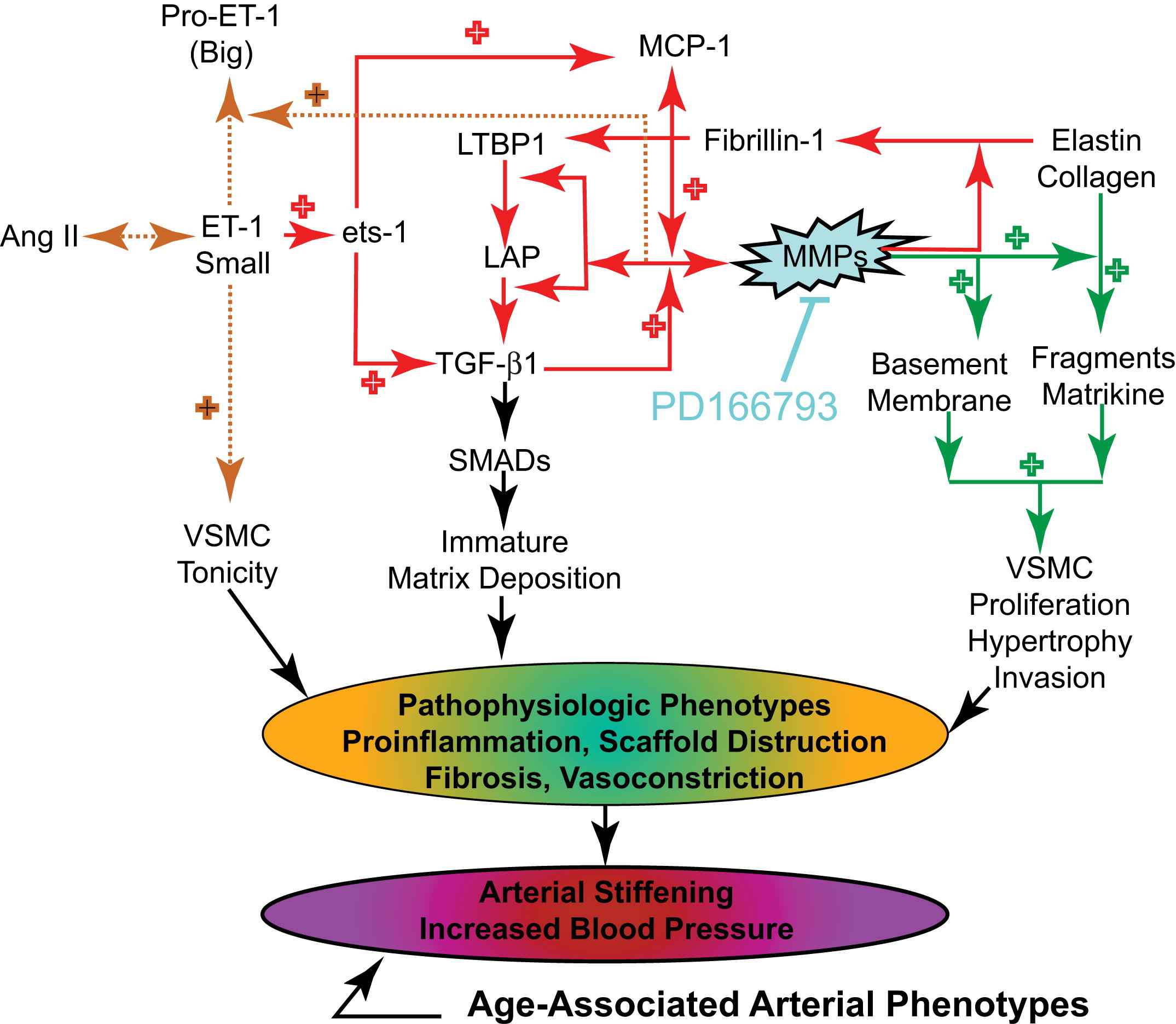
Key roles of matrix metalloprotease in age-associated changes in aortic wall phenotypes.
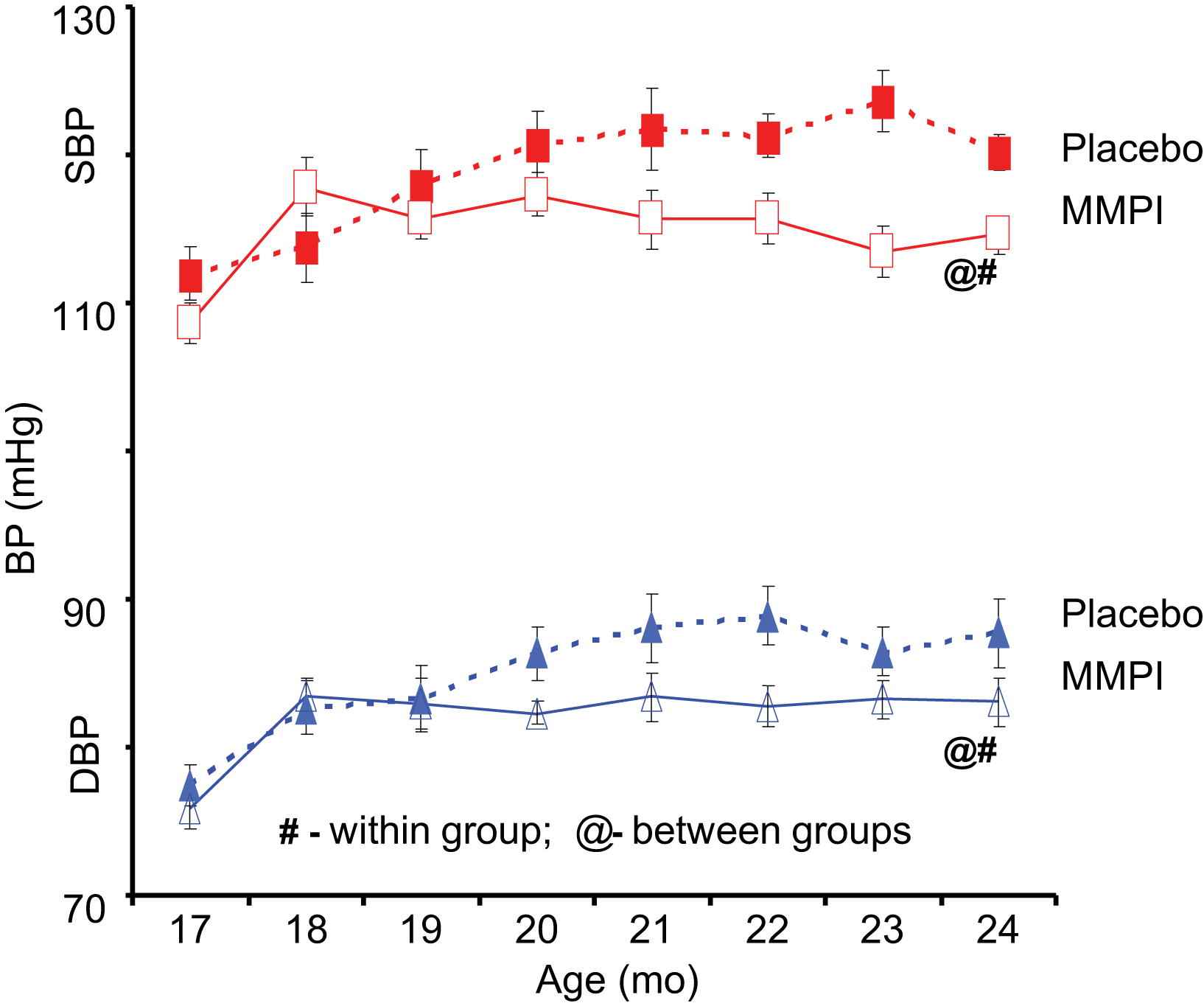
Chronic Matrix Metalloprotease Inhibition (MMPI) prevents an age-associated increase in arterial pressure in rats.
Acute exposure of cultured vascular smooth muscle cells in vitro to proendothelin 1 increased both the transcription and translation of ets-1, and these effects were markedly reduced by MMP inhibition. Furthermore, infection of vascular smooth muscle cells with an adenovirus harboring a full-length ets-1 cDNA increased activities of both transforming growth factor-β1 and monocyte chemoattractant protein 1. Collectively, these results indicate that MMP inhibition retards age-associated arterial proinflammatory signaling, and this is accompanied by preservation of intact elastin fibers, a reduction in collagen, and blunting of an age-associated increase in blood pressure.
Intertwining impacts of age, hypertension, early atherosclerosis and diabetes on the arterial wall
“Aging”-associated arterial changes and those associated with hypertension (and early atherosclerosis and diabetes) are fundamentally intertwined at the cellular and molecular levels.
A Megacept emerges with the realization that in arteries of younger animals, in response to experimental induction of hypertension or early atherosclerosis or diabetes, parts of this proinflammatory profile within the arterial wall that have been studied to date are strikingly similar to the profile that occurs with advancing age1 (Fig. 10).
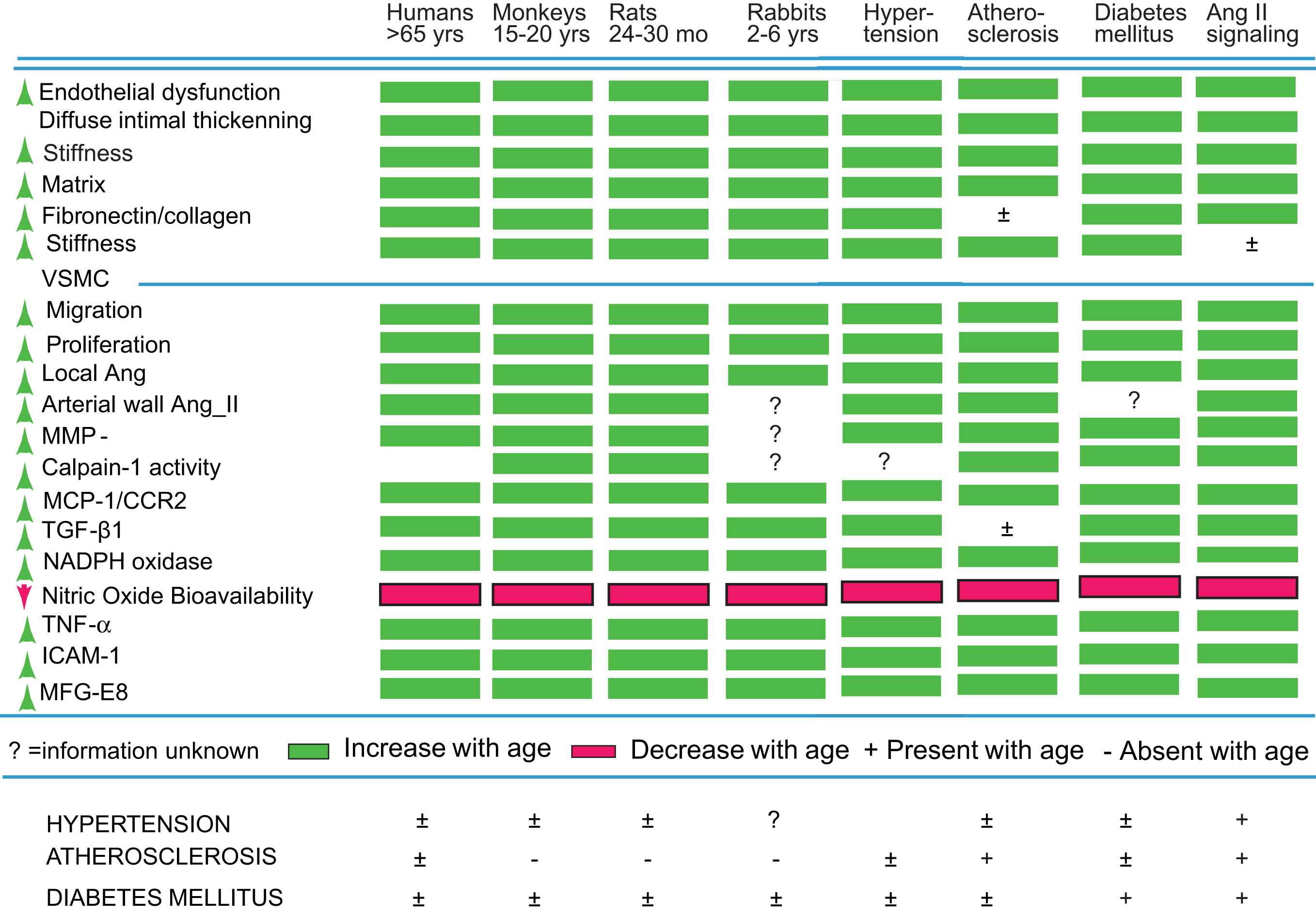
Chronic arterial inflammation and arterial remodeling during aging are strikingly similar to hypertension, atherosclerosis, and diabetes mellitus.
In summary, “aging”-associated arterial changes and those associated with hypertension (and early atherosclerosis and diabetes) are fundamentally intertwined at the cellular and molecular levels. In humans, other well-known risk factors (e.g., excess food intake, altered dietary lipid and metabolism, smoking, and lack of exercise) likely interact with this arterial substrate that has been altered during aging, and that renders the aging artery a “fertile soil” that facilitates the initiation and progression of these arterial diseases. Some lifestyle and pharmacologic interventions have already proved to be effective in preventing or ameliorating hypertension associated with aging. The cellular/molecular proinflammatory mechanisms that underlie arterial aging are novel putative candidates to be targeted by interventions aimed at attenuating arterial aging, and thus possibly attenuating the major risk factor for hypertension and atherosclerosis.
References
Cite this article
TY - JOUR AU - Edward G. Lakatta PY - 2013 DA - 2013/03/13 TI - The reality of aging viewed from the arterial wall☆ JO - Artery Research SP - 73 EP - 80 VL - 7 IS - 2 SN - 1876-4401 UR - https://doi.org/10.1016/j.artres.2013.01.003 DO - 10.1016/j.artres.2013.01.003 ID - Lakatta2013 ER -